Running Head: COPD Genetics
Funding: K12 HL120004, The Sheila J. Goodnight, MD, FCCP, Clinical Research Grant in Women’s Lung Health, R01HL089856; P01HL105339; R01HL075478
Date of Acceptance: February 28, 2014
Abbreviations: single nucleotide polymorphism, SNP; genome-wide association studies, GWAS; forced expiratory volume in 1 second, FEV1; early-onset COPD, EOCOPD; National Emphysema Treatment Trial, NETT; Normative Aging Study, NAS; International COPD Genetics Network, ICGN; linkage disequilibrium, LD; Evaluation of COPD Longitudinally to Identify Surrogate Endpoints, ECLIPSE; cigarettes per day, CPD; forced volume capacity, FVC; Cohorts for Heart and Aging Research in Genetic Epidemiology Consortium, CHARGE; messenger RNA, mRNA; body mass index, BMI; C-reactive protein, CRP; tumor necrosis factor-alpha, TNF-alpha; Clara cell secretory protein, CC16; surfactant protein D, SP-D;
Citation: Hardin M, Silverman EK. Chronic obstructive pulmonary disease genetics: a review of the past and a look into the future. Chronic Obstr Pulm Dis. 2014; 1(1): 33-46. doi: http://doi.org/10.15326/jcopdf.1.1.2014.0120
Introduction
Chronic obstructive pulmonary disease (COPD) is a complex disorder that results from both environmental and genetic risk factors. Although cigarette smoking is the greatest environmental risk factor for COPD, not all smokers develop COPD and lung function decline among smokers is highly variable. Early familial aggregation and linkage analysis studies strongly suggested genetic contributions to COPD, and recent genome-wide association studies have identified several genomic regions that are clearly related to COPD susceptibility. However, despite recent advances in COPD genetics, much of the heritability of COPD remains unexplained. The genetic determinants of COPD are likely composed of multiple genetic susceptibility variants of modest effect size and/or rare variants of large effect, acting in concert to create a diverse array of COPD-related phenotypes.
Unlike monogenic disorders such as cystic fibrosis, we are only beginning to understand the genetics of complex diseases like COPD. Despite initial advances that early on identified the serpin peptidase inhibitor, clade A (alpha-1 antiproteinase, antitrypsin), member 1 gene, SERPINA1, as responsible for the increased risk of pulmonary and hepatic disease in severe alpha-1 antitrypsin deficiency, determining the complex genetic system that results in other types of COPD has proven more difficult. Linkage analysis studies in families highlighted broad areas of the genome that were potentially involved in COPD pathogenesis. Candidate gene association analyses, using a priori hypotheses regarding COPD genetics, were initially promising; however, many of these findings were not replicated in additional populations.2
With the use of high throughput, single nucleotide polymorphism (SNP) genotyping array panels, researchers are now able to interrogate hundreds of thousands of SNPs in genome-wide association studies. These large-scale studies have revealed multiple promising regions of the genome for COPD susceptibility. However, COPD is likely the result of common variants of modest effect size and rare variants that may have a range of effect sizes. Studies capable of finding the rare variants will typically require DNA resequencing or dense genotyping panels; if the rare variant effects are not great, extremely large sample sizes may be required. Association studies, including genome-wide association studies (GWAS), can detect statistical relationships of genetic variants to disease phenotypes, but they do not identify the function behind these associations. Several recent advances in GWAS approaches have begun to address these limitations, including the ability to impute ungenotyped SNPs accurately as well as the development of large collaborative consortia. Faster and inexpensive whole-genome sequencing will enable fine-mapping and improved identification of rare variants. Additionally, refining phenotypic descriptions of COPD, by employing both clinical and hypothesis-free machine-learning techniques to distinguish COPD subtypes, may lead to more accurate clinical endpoints that can be used to study genetic associations. These analyses will also require a greater appreciation for how race and sex may impact COPD development. Finally, moving beyond the GWAS era, the emerging field of network medicine provides the tools to create functional models of genetic and environmental disease pathways.3
This review will briefly describe pre-GWAS genetic studies of COPD, then describe current COPD GWAS including those aimed at determining lung function genes as well as other COPD-related phenotypes. Finally, we will discuss the role of genetic studies in providing key input for integrative functional approaches to develop complex networks relevant to disease pathophysiology and treatment.
Familial Aggregation , Linkage, and Candidate Gene Analyses
Twin studies have demonstrated significant heritability of lung function levels,4,5 and suggested that as much as 60% of individual susceptibility to COPD could be explained by genetic factors.6 Familial aggregation studies compare the risk of disease in relatives of affected individuals to the risk for the general population. An increased risk can strongly suggest a genetic component to disease. For example, Cohen and colleagues demonstrated pulmonary dysfunction among relatives of COPD probands that was independent of smoking, gender, race and socioeconomic status.7 Silverman and colleagues developed a family-based cohort of individuals with severe, early-onset COPD (Boston Early-Onset COPD Study). Using this population, they demonstrated that current or ex-smoking first-degree relatives of probands with severe, early-onset COPD had an increased risk for reduced forced expiratory volume in one second (FEV1).8
The Boston Early-Onset COPD (EOCOPD) Study was also used for linkage analysis studies. In these studies, investigators harnessed knowledge of genetic markers and recombination events in family-based genetic studies to identify genomic regions linked to disease. Although such complex disease studies provide evidence for genetic linkage, they typically identify large genomic regions, millions of base pairs in length, that contain the region of interest. Early linkage studies in COPD provided evidence for linkage between regions on chromosomes 1, 2, 12, and 19 with COPD.8-10
In addition to family-based linkage studies, multiple investigators have used biologic hypotheses to select candidate genes for their association with COPD. In these studies, investigators choose SNPs from a gene or region of interest that is suggestive of a role in disease. Over 100 candidate gene studies in COPD have been published with many regions identified as potential COPD loci. However, most of these regions have not been able to be reproduced in subsequent studies. Significance thresholds have sometimes been adjusted for multiple testing within the study, but candidate gene associations have rarely met genome-wide levels of statistical significance—typically p < 5 x 10-8.
Several candidate genes have demonstrated associations that appear to be valid, either through biological plausibility or replication in multiple datasets. Although individuals with PI ZZ genotype and severe alpha-1 antitrypsin deficiency are clearly at increased risk for COPD, it was not known whether heterozygous PI MZ gene individuals, with moderate decreases in circulating alpha-1 antitrypsin levels, would also be at risk. Sorheim and colleagues demonstrated in both a case-control and family-based population that PI MZ individuals are at increased risk for reduced lung function as well as increased emphysema on CT scan imaging.11 Recently, Molloy and colleagues found that PI MZ relatives of COPD cases who smoke cigarettes have increased risk for COPD compared to PI MM smokers, while PI MZ nonsmokers did not have increased risk for COPD—suggesting a gene-by-smoking interaction.12
Hunninghake, Cho and colleagues successfully identified genetic markers in the matrix metalloproteinase-12, MMP12, gene as associated with lung function and COPD in high risk populations, using a candidate gene approach encompassing diverse study populations.13 It was known that increased MMP12 expression led to elastin degradation, 14,15 increased expression was seen in the alveoli of human smokers compared to nonsmokers,16 and knock-out mice did not develop emphysema in response to cigarette smoke. 17 These researchers investigated diverse cohorts with lung disease, including 3 cohorts with childhood asthma and 4 adult cohorts with COPD. They tested SNPs in or near the MMP12 gene for association with pre-bronchodilator FEV1 in all cohorts, as well as COPD affection status in 2 COPD populations. They found that a variant in the promoter region of MMP12 (rs2276109) was protective for lung function in both children with asthma and adult smokers, strongly suggesting a role for this gene in lung function at multiple stages of life.
However, while candidate gene studies were initially promising, many of the findings have not been replicated in subsequent analyses. This is usually because, unlike the MMP12 example, these associations were initially determined in small cohorts with variable definitions of COPD. Smolonska and colleagues performed a meta-analysis of candidate gene studies that were specifically selected for their well-defined phenotype as well as biologically plausible candidate gene associations. Many promising candidates were no longer significant when combined in a meta-analysis. In fact, after examining 69 studies with positive genetic associations for 20 polymorphisms in 12 genes, they found that only 3 polymorphisms in the transforming growth factor beta -1 gene, TGFB1 were significant in their meta-analysis. However, they cautioned interpretation even of this finding, given the small size of the cohorts in which it was found. In addition, they noted the importance of considering ethnicity in constructing candidate gene analyses, as many findings were able to be replicated in one ethnicity but not others.18
In a similar meta-analysis, Castaldi and colleagues examined all candidate gene studies in COPD performed up to 2009.19 The definition of COPD varied significantly in many studies, from strict spirometric definitions to physician diagnoses. They were able to identify 27 variants that were studied in 3 or more independent populations. In a meta-analysis, they identified nominal statistical significance for variants in 4 genes (glutathione S-transferase mu 1 gene, GSTM1, tumor necrosis factor (TNF), beta-1 gene, TGFB1, and superoxide dismutase 3 gene, SOD3). After sensitivity analysis, only the GSTM1 locus, out of 100 potential associations, remained robust.
These studies highlight the limited power of candidate gene analyses to identify genetic variants associated with COPD. However, there have been several integrative analyses that have identified novel associations by combining candidate gene analysis with additional genomic approaches that have identified more robust associations. These studies have additionally incorporated functional analyses as well as replication populations and therefore appear more promising.
Fine mapping, performed by genotyping many SNPs in chromosomal regions previously identified through linkage studies, has also helped to clarify genes of interest located in these regions. Hersh and colleagues used fine mapping techniques in a region on chromosome arm 2q previously identified through linkage studies, to identify X-ray repair complementing defective repair in Chinese hamster cells (double-strand-break repairing) gene, XRCC5, as associated with COPD, using information from the National Emphysema Treatment Trial (NETT)- Normative Aging Study (NAS) and GenKOLS and followed up in the family-based International COPD Genetics Network (ICGN) and Boston EOCOPD studies.20 In a different analysis, Hersh and colleagues examined a region on chromosome 12p that had been previously related to COPD susceptibility through linkage analysis. As part of their analysis, they genotyped 1387 SNPs in 386 individuals from the NETT-NAS cohort to identify a panel of SNPs that were significantly associated with COPD. Twenty-six of these SNPs were then tested for replication in the Boston EOCOPD Study. They found a variant in the SRY (Sex-determining region Y)-Box5 gene, SOX5, that was significantly associated with COPD in both studies, but not in a third family-based cohort. They additionally demonstrated decreased SOX5 expression in lung tissue from individuals with COPD compared to controls that correlated with lung function. SOX5 knock-out and heterozygous mouse models demonstrated changes in early lung development. By incorporating functional genetic analysis, Hersh and colleagues provided increased evidence for a role of SOX5 in COPD pathogenesis.
COPD Genome-Wide Association Studies
To date, COPD genome-wide association studies have been underpowered to detect variants of modest effect size. In contrast, population-based studies examining variants associated with spirometric values in larger cohorts with a broad range of pulmonary function have been able to identify multiple genetic variants that are clearly associated with pulmonary function. Despite these challenges, GWAS have identified several regions that appear to be associated with COPD case-control status. With the development of larger, better powered cohorts with genotype information, more COPD-related genetic variants may be identified in the future.
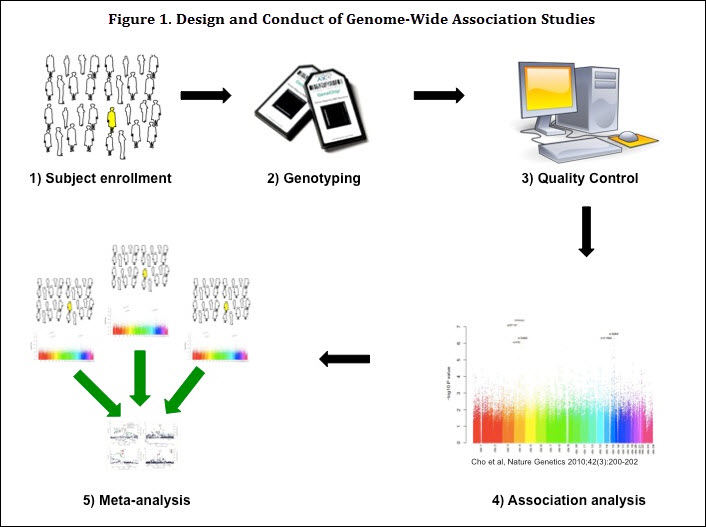 With the advent of high-throughput genotyping techniques, investigators are now able to perform genome-wide SNP-based association testing, GWAS, in a relatively inexpensive and rapid fashion (Figure 1). These tests allow investigators to examine a genome-wide panel of SNPs for association with a phenotype of interest, such as the presence or absence of COPD. This is in contrast to candidate gene studies that require a priori hypotheses for genes that may be associated with COPD. GWAS have therefore allowed recognition of novel genomic regions that would not previously have been considered as part of COPD pathogenesis.
With the advent of high-throughput genotyping techniques, investigators are now able to perform genome-wide SNP-based association testing, GWAS, in a relatively inexpensive and rapid fashion (Figure 1). These tests allow investigators to examine a genome-wide panel of SNPs for association with a phenotype of interest, such as the presence or absence of COPD. This is in contrast to candidate gene studies that require a priori hypotheses for genes that may be associated with COPD. GWAS have therefore allowed recognition of novel genomic regions that would not previously have been considered as part of COPD pathogenesis.
The genotype of the SNP is determined through rapid multiplex screening. These genotypes are tested for association with the phenotype using linear or logistic regression techniques. Each study tests for association with hundreds of thousands of SNPs, and therefore, the significance level must be adjusted for these multiple tests. The standard method is to use a Bonferroni correction of alpha = 0.05 divided by the number of SNPs tested. Currently a P value of less than 5x10-8 is considered necessary for genome-wide significance.21 Investigators have additionally harvested the knowledge of the sequenced human genome as well as linkage disequilibrium (LD) patterns to impute SNPs that are not genotyped. In this manner, investigators can determine predicted SNP genotypes with certain levels of probability for millions more SNPs that were not even genotyped, thus increasing the power of the study to determine a significant association. Because these tests are subject to multiple testing from incorporating so many SNPs, the standard currently is to then attempt replication of identified loci in additional populations.
However, despite advances in genotyping techniques, only a few loci have been identified to be associated with COPD affection status at genome-wide significance. COPD is a complex disease with many disease-related phenotypes and likely multiple component subtypes. Many variants that do not reach strict genome-wide significance thresholds likely play some role in disease processes. In order to identify COPD genetic determinants, it will be necessary not just to increase the number of SNPs being studied, but more importantly, to increase the sample size. These studies require very large cohorts that are usually the result of multiple investigators from many sites working together. Although variants associated with common disease may occur frequently in the population, their effect sizes are typically modest. In comparison to monogenic traits like cystic fibrosis or oligogenic traits like age-related macular degeneration, COPD variants likely have odds ratios for disease risk less than 1.5. Therefore, large study populations are 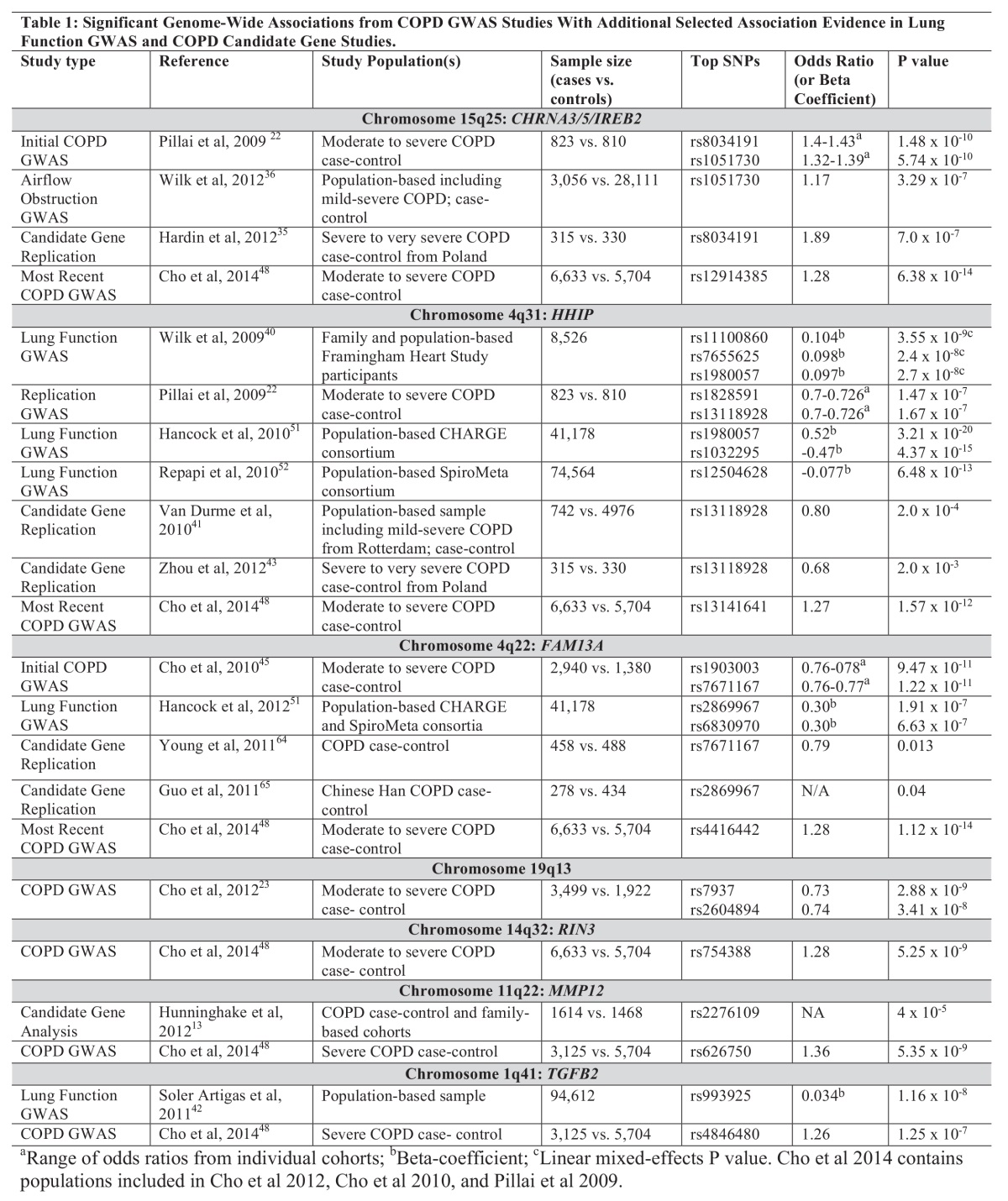 necessary to obtain the power to identify these associations. As will be seen below, COPD researchers are pooling resources to obtain collaborative genetic studies composed of multiple case-control COPD cohorts that have genetic information (Table 1). By increasing the number of participating cohorts with genetic information, new variants may be discovered. Additionally, in order to truly capture the association, investigators will need to refine the COPD phenotypes that are being used as the outcome. For example, COPD characterized by upper-lobe predominant emphysema may be very different from COPD with significant lung function changes but no emphysematous changes.
necessary to obtain the power to identify these associations. As will be seen below, COPD researchers are pooling resources to obtain collaborative genetic studies composed of multiple case-control COPD cohorts that have genetic information (Table 1). By increasing the number of participating cohorts with genetic information, new variants may be discovered. Additionally, in order to truly capture the association, investigators will need to refine the COPD phenotypes that are being used as the outcome. For example, COPD characterized by upper-lobe predominant emphysema may be very different from COPD with significant lung function changes but no emphysematous changes.
Chromosome Band 15q25: IREB2 and CHRNA3/5
The chromosome band 15q25.1 is an intriguing genomic region for COPD susceptibility. There are several genes in this region that have been identified as likely having a role in COPD, including the cholinergic receptor, nicotinic, alpha 3 (neuronal) gene, CHRNA3, the cholinergic receptor, nicotinic, alpha 5 (neuronal) gene, CHRNA5, and the iron-responsive element binding protein 2 gene, IREB2. GWAS and integrative genomics approaches have identified both CHRNA3/5 and IREB2 as potential candidates for a role in COPD susceptibility and progression. Interestingly, despite their close genomic proximity, it appears that these genes have very different roles in COPD pathogenesis. This particularly gene-dense region highlights some of the limitations of GWAS studies, as the strong association with one marker may cloud interpretation of results.
Furthermore, the CHRNA3/5 region has additionally been associated with lung cancer, peripheral arterial disease, and nicotine addiction. GWAS are limited in their ability to distinguish a role for CHRNA3/5 in COPD pathogenesis independent from nicotine addiction.
CHRNA3/5 and COPD
Pillai and colleagues used a multi-stage replication approach to identify a region on chromosome 15 associated with COPD.22 They performed an initial GWAS study in a case-control population of current and former smokers from Norway(GenKOLS) that included 823 individuals with COPD and 810 controls. They then took the top 100 SNPs with the lowest P values from this cohort and tested them for association in an additional, family-based cohort, ICGN. Seven of these SNPs were nominally significant with a P value of < 0.05 and the same direction of effect as the first study. These SNPs were then tested for association in 2 other studies including severe COPD cases and controls from NETT/NAS and families with EOCOPD. These latter cohorts are distinct from the former because of a greater severity of COPD (NETT), as well as an earlier onset of COPD (EOCOPD). The authors found a consistent significant association between COPD and 2 SNPs in or near the CHRNA3/5 gene locus. In a subsequent pooled meta-analysis including the GenKOLS, Evaluation of COPD Longitudinally to Identify Surrogate Endpoints (ECLIPSE) and NETT/NAS cohorts, as well as the first 1000 individuals from the COPDGene® study, variants in LD with their top SNP demonstrated significant association with COPD, even after adjusting for pack years of smoking, strongly suggesting a role for this locus in COPD pathogenesis.23
CHRNA3/5 and Nicotine Addiction
The CHRNA3/5 region encodes several nicotinic acetylcholine receptor subunit genes. This region had previously been associated with smoking behavior as well as lung cancer and peripheral arterial disease.24-26 In a series of concurrently published meta-analyses, several large cohorts investigating the association of smoking-related traits with the 15q25 region demonstrated significant associations with variants in this region and measurements of nicotine addiction and smoking quantity.27-29 Saccone and colleagues identified two distinct haplotypes in this region that were associated with heavy smoking behavior from a meta-analysis that included 34 different studies. Their most significant haplotype contained SNPs in LD with those previously associated with COPD. However, they only identified nominal association between this haplotype and COPD risk using a small subset of their cohorts with known COPD case/control status.30
Although variants in this region do appear to impact nicotine addiction and smoking-related behaviors, it remains possible that COPD-related variants confer additional COPD susceptibility. This was suggested in a recent investigation by Siedlinski, et al. The authors used mediation analysis to attempt to distinguish associations between the CHRNA3/5 locus (rs1051730) and the IREB2 locus (rs13180) (discussed below) and COPD that were independent from smoking behavior. They found that carriers of the rs1051730 variant showed increased odds of developing COPD across tertiles of smoking intensity, indicating that smoking exposure did impact the effect of this SNP on COPD susceptibility. However, they also found significant independent effects of this SNP on COPD,31 suggesting in fact that the CHRNA3/5 locus may also play a role in COPD development independent from impacting smoking behavior.
IREB2
The CHRNA3/5 locus is in close proximity to IREB2, a gene that has also been associated with COPD. IREB2 encodes the iron response protein 2 (IRP2), which plays a role in cellular iron metabolism, and may be active at lower oxygen tensions.32 The IREB2 locus was first associated with COPD through a series of genomic and genetic approaches by DeMeo and colleagues.33 In this study, the authors identified candidate regions by using microarray gene expression analysis of lung tissue to identify differentially expressed genes between COPD cases and controls. These regions were used to identify candidate SNPs for association analyses with COPD and FEV1. They identified SNPs in the IREB2 gene that were associated with COPD affection status as well as FEV1. In addition, they demonstrated increased IREB2 expression in lung tissue from COPD cases compared to controls, and demonstrated IREB2 protein localization in airway epithelial, endothelial, smooth muscle cells, and macrophages. This study demonstrated the utility of integrating genomic information to guide genetic association testing, and provided some functional explanation for a role of IREB2 in COPD pathogenesis.
The association between IREB2 and COPD was replicated in a European case/control study in which investigators demonstrated association between COPD and 3 SNPs representing 3 distinct haplotypes in IREB2.34 The SNP that was most significantly associated with COPD in DeMeo’s paper demonstrated significant association with moderate to severe COPD in a Polish cohort.35 In their mediation analysis, Siedlinski demonstrated that IREB2 influenced COPD susceptibility independently of cigarette smoke exposure.31
In a population-based meta-analysis that included 32,875 cases,Wilk and colleagues found significant association between SNPs from the 15q25 chromosome band and smokers with airflow obstruction. Among never-smokers with obstruction, they found nominal association between variants in both the CHRNA5 and CHRNA3 genes and airflow obstruction. They additionally demonstrated expression of these genes in whole lung samples.36
The 15q25 band appears to encode several genes that are involved in COPD pathogenesis. The CHRNA3/5 locus may act partially through impacting nicotine addiction or cigarette exposure, but variants in this region additionally may impact COPD development independently. Variants in the IREB2 locus appear to confer COPD risk independent of smoking behavior, likely through an impact on iron metabolism.
HHIP
Many GWAS associations are located within non-coding regions of genes. SNPs identified in intergenic loci may also play a functional role, most likely by influencing gene regulation. Researchers have identified a locus upstream of the hedgehog interacting protein gene, HHIP, on chromosome band 4q31, that consistently replicates in COPD genetic association studies. The HHIP protein product is essential for lung development37, 38 ; by binding to the sonic hedgehog protein, it can activate the hedgehog signaling pathway.39 Mice with homozygous deletion of the HHIP gene are not viable, due to defects in lung development.38
Wilk and colleagues first identified genetic variants near the HHIP gene as associated with lung function at genome-wide significance in a population-based cohort.40 Pillai and colleagues, in their previously mentioned COPD GWAS analysis, additionally demonstrated that 2 SNPs (rs1828591 and rs13118928) in the HHIP locus demonstrated consistent association with COPD in 3 cohorts. Although not genome-wide significant, with P values 1.47 x 10-7 and 1.67 x 10-7, the effect size was in the same direction in all 3 cohorts, suggesting a likely association that the study was underpowered to demonstrate. This finding was subsequently replicated in a population-based cohort including 742 cases with COPD41 as well as with a different SNP near the HHIP locus in a candidate gene study.42
Zhou and colleagues demonstrated a potential role for the HHIP locus in COPD pathogenesis through a series of functional analyses, integrating genetic and molecular biology techniques.43 They first demonstrated a significant association between the previously identified upstream variant and severe COPD in a cohort of Caucasian smokers from Poland. They then demonstrated reduced HHIP expression in the lung tissue of individuals with COPD compared to smoking controls. In order to further determine if the functional variant was located within the HHIP gene, they resequenced both the upstream region, the intervening region, and the exons and introns of HHIP in 29 individuals with severe EOCOPD. They found 21 SNPs in the upstream region previously identified, but no common, nonsynonymous variants in the HHIP gene itself, suggesting that the association was in fact identifying cis-acting regulatory variants in the upstream region that were involved in COPD pathogenesis.
In order to better determine a functional role for genetic variants in this region, they performed a series of functional tests. They demonstrated that this region contained a long-range enhancer for the HHIP promoter using chromosome conformation capture (3C) assays in bronchial epithelial and fetal lung fibroblast cell lines. They demonstrated that the risk haplotype for COPD was associated with decreased HHIP promoter activity. Finally, they identified the risk site as a potential binding site for Sp3, a transcription factor which often serves as a transcriptional repressor. The risk allele demonstrated stronger Sp3 binding, potentially leading to decreased expression of HHIP. These findings strengthened our understanding of HHIP as having a potential role in COPD pathogenesis, with variants identified through GWAS as potentially down-regulating HHIP expression and increasing COPD risk. In a subsequent analysis, Zhou and colleagues used human bronchial epithelial cells to identify differential gene expression in the setting of HHIP silencing to identify potential targets for the role of HHIP in COPD pathogenesis. Integrating expression microarray, functional annotation, and network science approaches, they identified extracellular matrix and cell profliferation pathways targeted by HHIP in COPD lung tissue.44
FAM13A
Cho and colleagues used genome-wide SNP genotyping data from 3 COPD cohorts that were used in the identification of the CHRNA3/CHRNA5/IREB2 and HHIP loci into a meta-analysis as well as a mega-analysis, creating one dataset for genome-wide analysis, containing 2940 cases and 1380 controls.45 In their mega-analysis, they identified 2 SNPs (rs1903003 and rs7671167) in linkage disequilibrium that were significantly associated with COPD (7.18 x 10-8 and 8.59 x 10-9 respectively). These SNPs are located on chromosome band 4q22.1 within the family with sequence similarity 13, member A gene, FAM13A.
They then attempted to replicate these associations in 3 additional cohorts, 502 cases and 504 controls from the COPDGene® cohort as well as the 2 family-based ICGN and Boston EOCOPD cohorts. These SNPs were significantly associated with COPD in the COPDGene® and ICGN cohorts, but not the Boston EOCOPD, possibly reflecting the different subject recruitment for the latter with severe and early-onset COPD. They did not identify any association or interaction between these SNPs and pack years of smoking. When they combined the results of all cohorts studied, the FAM13A locus demonstrated a genome-wide significant association with COPD with P=1.22 x 10-11.
The identified variants are located downstream of a Rho GTPase-activating domain within the FAM13A gene and are likely involved in signal transduction. FAM13A expression has been shown to increase in tissue in response to hypoxemia.46 However, a potential mechanism for the role of this gene in COPD pathogenesis is not currently known.
Chromosome Band 19q13
Cho and colleagues combined the genetic information of 4 cohorts, ECLIPSE, NETT/NAS, GenKOLS and initial genetic data on 1,000 individuals from the COPDGene study to perform a COPD case-control meta-analysis.23 They imputed missing genotypes in each cohort against the 1000 Genomes Project data, allowing better combination of the genetic information from each cohort. Along with previously identified regions (CHRNA3/5/IREB2, HHIP and FAM13A), they additionally identified variants from a novel region on 19q13 as significantly associated with COPD. These variants, rs7937 at P=2.88 x 10-9 and rs2604894 at P=3.41 x 10-8, demonstrated genome-wide significance in a meta-analysis of these cohorts. These same loci had previously been identified as associated with cigarettes per day (CPD) in a smoking behavior GWAS.29 However, in Cho’s analysis, these variants did not demonstrate association with CPD or pack years. In a replication population from the family-based ICGN cohort, the 19q13 SNPs were not associated with COPD status, but showed some evidence for association with severe COPD (p=0.09 and 0.017) as well as pre-bronchodilator FEV1 (P=0.08 and 0.04).
The 19q13 genomic region contains several genes that could potentially be involved in COPD pathogenesis. Variants of the cytochrome P450, family 2, subfamily A, polypeptide 6 gene, CYP2A6, have been associated with smoking behavior 28,29 and lung cancer.24 The other genes in this region include the RAB4B, member RASoncogene family gene, RAB4B, the melanoma inhibitory activity gene, MIA, and the egl-9 family hypoxia-inducible factor 2 gene, EGLN2. Recent gene expression studies of airway basal cells in smokers demonstrate increased gene expression for several genes expressed in this region, including EGLN2.47 As this locus has previously demonstrated strong associations with smoking behavior, it is likely that its role in COPD pathogenesis is through smoking; however, additional mechanisms responsible for this association cannot be ruled out.
COPD Affection Status Meta-Analysis
A recent study which included genome-wide SNP genotyping in the entire COPDGene® cohort along with meta-analysis of multiple studies performed by Cho and colleagues 48 confirmed the prior associations with HHIP, CHRNA3/5/IREB2, and FAM13A. In addition, this analysis identified new associations with moderate-severe COPD near the Rab5 GTPase binding protein gene, RIN3, and with severe COPD near MMP12 and the transforming growth factor beta 2 gene, TGFB2. They combined COPDGene® genotyping data with the NETT/NAS, ECLIPSE, and GenKOLS populations in a COPD affection status meta-analysis. They used the family-based ICGN population for replication of their novel findings. In addition to identifying additional genes associated with COPD, this analysis demonstrated that larger COPD populations can indeed identify novel COPD risk genes. In addition, in their stratified analysis including only severe COPD individuals, they identified both novel markers not seen in the whole population, as well as significantly stronger effect sizes for the known associations. This latter finding suggests that dividing heterogeneous COPD individuals into more phenotypically distinct groups, such as severe COPD, may be more likely to identify genetic associations.
Expression Quantitative Trait Analyses
Genome-wide SNP data can be used with gene expression data to identify potential functional roles for GWAS associations. Qiu and colleagues integrated SNP and genome-wide gene expression microarray data to identify expression quantitative trait loci (eQTLs) in sputum from a subset of individuals from the ECLIPSE study.49 They identified cis-eQTL SNPs on chromosome 15 in IREB2 and CHRNA5 as being associated with COPD in a combined GWAS including ECLIPSE, GenKOLS and NETT-NAS participants. In addition, they identified a novel locus on chromosome 6 in the psoriasis susceptibility 1 candidate 1 gene, PSORS1C1. This SNP was in the top 74 most significant SNPs in their primary analysis and was significantly associated with COPD in the ICGN replication population, although not in COPDGene®. Their findings identified previous GWAS variants on IREB2 and CHRNA3/5 as potentially involved in affecting gene expression. They examined eQTLs in HHIP and FAM13A and did not find association with COPD. They concluded that this could indicate these variants may not be involved in sputum gene expression, but they could impact gene expression in other tissues. Other researchers have identified eQTL SNPs for HHIP, EGLN2 and FAM13A associated with gene expression by combining genotypes and lung tissue expression data.50
Lung Function Genome-Wide Association Studies
In addition to COPD GWAS, several investigative groups have pursued genetic associations with measurements of lung function levels in population-based samples. As reduction in lung function, especially FEV1/forced volume capacity (FVC), is a hallmark of COPD, it would seem reasonable that variants identified in these studies could additionally be involved in COPD pathogenesis. These investigations may have greater power to detect associated variants because they are examining a continuous, rather than dichotomous trait. They also use larger, population-based samples rather than those defined by the presence or absence of COPD. These studies have revealed a large number of genetic loci associated with FEV1 and FEV1/FVC, and in some cases have confirmed loci associated with COPD. These lung function variants, in turn, make strong candidates for testing for association with COPD in disease-based cohorts. It should be noted, however, that these are population-based samples with only minimal inclusion of individuals with COPD, and many individuals were not current or former smokers.
Wilk and colleagues performed the first GWAS of lung function with significant results.40 In their analysis, discussed previously, they included 7,691 family-based participants from the Framingham Heart Study and examined genotyped and imputed SNPs for association with FEV1/FVC percent predicted. Twenty-seven SNPs reached genome-wide significance, and these SNPs were located in an intergenic region near the HHIP gene. They then tested SNPs from the regions with the lowest P values for association with airflow obstruction in the Family Heart Study. This cohort of 835 white individuals included 225 participants with airflow obstruction as well as 610 controls. The HHIP region was the only region that was significantly associated with FEV1/FVC percent predicted. This region was additionally significantly associated with FEV1 as well as a dichotomous variable for airflow obstruction.
This analysis was followed by 2 larger consortium studies. Hancock and colleagues performed a GWAS of FEV1 and FEV1/FVC in 20,890 individuals from 4 cohorts included in the Cohorts for Heart and Aging Research in Genetic Epidemiology Consortium (CHARGE).51 This population-based study included current and former smokers as well as non-smokers. They additionally used the SpiroMeta consortium as a replication population. They identified 8 loci significantly associated with FEV1/FVC, including HHIP and FAM13A as well as GPR126,ADAM19, AGER/PPT2, PTCH1, PID1 and HTR4 and one locus associated with FEV1 (INTS12/GSTCD/NPNT). Repapi and colleagues used 20,288 individuals from the SpiroMeta consortium to test associations with lung function measurements.52 They then replicated their top SNPs by genotyping in up to 32,184 individuals as well as in silico data from the CHARGE consortium and an additional 883 individuals from the HEALTH 2000 study. They identified variants in genes associated with FEV1 (GSTCD, HTR4 and the tensin1 gene, TNS1) as well as FEV1/FVC (AGER, and the thrombospondin, type I, domain containing 4 gene, THSD4). They confirmed the prior associations between the HHIP locus and FEV1 and FEV1/FVC. They additionally demonstrated messenger RNA (mRNA) expression of these associated genes in lung tissue.
These studies were followed by a larger meta-analysis of 23 studies in CHARGE and SpiroMeta that tested over 2.5 million genotyped and imputed SNPs in 48,201 white individuals.53 The SNPs with the lowest P values were then examined in 17 additional studies including 46,411 individuals. These authors replicated the prior lung function GWAS hits, and identified 1 new region associated with both FEV1 and FEV1/FVC (CDC123), 3 additional FEV1 regions (MECOM, ZKSCAN3/ZNF323, C10orf11) and 12 FEV1/FVC regions (MFAP2, TGFB2, HDAC4, RARB, SPATA9, NCR3, ARMC2, LRP1, CCDC38, MMP15, CFDP1, KCNE2).
Some of these lung function SNPs have subsequently been demonstrated to be associated with COPD in COPD case-control cohorts. Soler-Artigas and colleagues tested SNPs from 5 lung function loci including TNS1, GSTCD, HTR4, AGER, and THSD4 in a population-based cohort that included 3,284 individuals with spirometrically- defined COPD and 17,538 controls. TNS1, GSTCD and HTR4 were significantly associated with COPD in this population. Castaldi and colleagues tested the 32 SNPs in or near 17 genes previously identified as associated with lung function in the CHARGE and SpiroMeta consortia as well as imputed SNPs from these 17 genes in 4 case-control COPD cohorts (NETT/NAS, GenKOLS, ECLIPSE and the first 1000 participants from COPDGene®).54 They identified 3 loci that were associated with COPD, including 4q24 (FLJ20184/INTS12/GSTCD/NPNT), 6p21 (AGER and PPT2), and 5q33 (ADAM19). The ADCY2 locus was additionally found to be associated with COPD in a cohort from Poland with severe COPD.35
In a further refinement of the connection between lung function GWAS loci and COPD, Hansel and colleagues used a COPD cohort to identify markers associated with lung function decline in mild-moderate COPD, and were able to identify two regions on chromosomes 10 and 14 suggestive of association with this trait.55
COPD-Related Phenotype Genome-Wide Association Studies
COPD is a complex disease with a variety of clinical presentations. This heterogeneity of COPD phenotypes could play a role in the modest effect sizes of GWAS associations for COPD. By refining COPD phenotypes, several investigators have identified additional markers associated with COPD, and in doing so have helped to clarify a functional role for these markers.
Pulmonary emphysema is a core component of COPD, described by progressive destruction of distal airspaces. Emphysema has been associated with reduced lung function56 and greater exacerbation frequency.57 In the first GWAS to examine variants for their association with emphysema, Kong, Cho and colleagues identified a SNP in the bicaudal D homolog 1 (Drosophila) gene, BICD1, as significantly associated with severe emphysema as assessed visually by a radiologist (P = 4.8 x 10-8) and suggestively associated with the presence of emphysema (P = 5.2 x 10-7).58 In their analysis, they tested variants from the ECLIPSE, GenKOLS and NETT cohorts for association with both quantitative and qualitative measures of emphysema. Interestingly, variants associated with quantitative densitometric measures of emphysema were less significantly associated than radiologist assessments, possibly due to different CT scan techniques in each study. The BICD1 protein is involved in controlling dynein function, and has been associated with telomere length in leukocytes,59 suggesting that the role of BICD1 in emphysema could be related to shortened telomeres.
Pillai and colleagues used the data from the ECLIPSE study to investigate the associations of known GWAS COPD genetic loci with COPD-related phenotypes, including spirometry, smoke exposure, body mass index (BMI), fat-free body mass index, emphysema, airway wall thickness, COPD exacerbations, and BODE score.60 They used the family-based ICGN cohort as a replication population. They found that the CHRNA3/5 locus was associated with pack years, emphysema and airflow obstruction. IREB2 was associated with FEV1, and HHIP was associated with airflow obstruction as well as COPD exacerbations. The association between the CHRNA3/5 locus and both pack years and COPD phenotypes could indicate that this locus plays several roles in COPD development. In contrast, the lack of association with pack years at any of the other sites may indicate pathways that are independent of nicotine addiction.
Wan and colleagues examined associations with BMI in COPD populations. COPD-related cachexia is a phenotype that impacts a subset of COPD individuals and has a significant impact on mortality, and BMI is a clinically relevant COPD phenotype.61 They performed a meta-analysis of 3 COPD case-control cohorts including ECLIPSE, GenKOLS and NETT, and replicated their top findings in the first 1000 individuals of the COPDGene® cohort. In the meta-analysis, their most significant SNP was located within the fat mass and obesity-associated gene, FTO. The risk allele from this gene has previously been demonstrated to be associated with being overweight or obese. In their study, the risk allele was associated with greater BMI, fat-free mass index (FFMI), FEV1 percent predicted, FEV1/FVC and less overall emphysema. Their study revealed a potentially protective role for the FTO gene in COPD, similar to that previously found for lung cancer.
Circulating blood biomarkers can provide insight into disease pathophysiology and biomarker levels could additionally reflect different phenotypic presentations of disease. GWAS of biomarkers in disease could potentially identify genes associated with certain pathways in a more specific fashion than GWAS of disease affection status. Kim and colleagues performed a GWAS for 2 pneumoproteins (clara cell secretory protein, CC16, and surfactant protein D, SP-D) as well as 5 inflammatory biomarkers (fibrinogen, C-reactive protein (CRP), interleukin-6 (IL-6), IL-8, and tumor necrosis factor-alpha (TNF-alpha) in the ECLIPSE study.62 They identified 1 region of association with Clara cell secretory protein (CC16) levels in COPD individuals near the secretoglobin, family 1A, member 1 (uteroglobin) gene, SCGB1A1,which encodes CC16 and another region of association 20 million base pairs away on the same chromosome. SNPs near the surfactant protein D coding gene, SFTPD, were associated with surfactant protein D (SP-D) levels, but SNPs on 2 other chromosomes were significantly associated as well, suggesting that these other chromosomal regions encode trans-acting regulatory factors. In addition, several of the SNPs associated with CC16 levels were also associated with sputum mRNA expression levels of this protein. Finally, they tested their top SNPs for association with COPD affection status in ECLIPSE, GenKOLS, and NETT-NAS and found nominal association with several of the loci associated with CC16 or SP-D and COPD. Their study was the first to highlight associations with pneumoproteins as well as pneumoprotein gene expression, and additionally demonstrated the utility of studying genetics of biomarkers in order to identify potential genetic determinants of complex traits.
Conclusions:
COPD is a complex disease, and variants in the genetic code that are responsible for disease pathogenesis likely include both common variants with modest effect and rare variants, potentially with stronger effect. Both situations require large sample sizes and dense genotyping or resequencing for identification. In 2010, an international meeting of COPD genetics investigators convened to address the needs of current COPD genetics and consider future collaborations. They identified the need for larger sample sizes including multiple ethnic groups as well as more precise COPD phenotyping. Among existing COPD genetics studies, there are over 14,700 cases and 37,600 controls with genome-wide genotyping data that could be available for collaborative studies.63
In addition to building large consortia, it will be necessary for investigators to incorporate newer genetics techniques with existing GWAS data to identify additional genes of small to modest effect size. Fine mapping techniques and eQTL analysis can help to identify functional variants within previously identified regions. Next generation DNA sequencing of the whole exome and ultimately the whole genome may help to identify rare variants of large effect. Using more precise COPD phenotyping, such as quantitative CT analysis to assess lung parenchyma and airway remodeling, will allow for focused analyses on more specific patterns of COPD presentation. Finally, systems biology approaches enable the combination of multiple data streams and provide a more comprehensive understanding of the functional role of variants identified through GWAS.3
The future is bright for COPD genetics. With greater collaboration among research groups, the use of novel ‘omics technologies, and incorporation of more ethnicities, there is a promise of many new discoveries to come.