Running Head: Pathophysiology of Emphysema
Abbreviations: chronic obstructive pulmonary disease; COPD; extracellular matrix; ECM; mammalian target of rapamycin; mTOR; vascular endothelial growth factor, VEGF
Citation: Goldklang M, Stockley R. Pathophysiology of emphysema and implications. Chronic Obstr Pulm Dis. 2016; 3(1): 454-458. doi: http://doi.org/10.15326/jcopdf.3.1.2015.0175
Introduction
Lung injury in COPD is the result of many different pathogenic processes within the lung. There are a multitude of factors that influence disease susceptibility, initiation of injury and progression of disease. The inhalation of cigarette smoke causes a variety of pro-inflammatory and oxidative stress cascades to be activated within the lung, with resultant protease production and alveolar cell apoptosis, all leading to lung destruction. This session provided an overview of key aspects of smoke-induced lung injury in order to better understand potential targets for new therapies.
Overview of Lung Injury in COPD: Types and Key Questions
Lung injury in emphysema is a result of inflammatory and destructive processes in response to cigarette smoke exposure. In patients with chronic obstructive pulmonary disease (COPD), pro-inflammatory and pro-destructive pathways are activated, at times independent of smoke exposure, and other anti-inflammatory, anti-oxidant, or repair pathways are down-regulated, all resulting in lung destruction. It has been long accepted that cigarette smoke leads to airway inflammation, but cigarette smoke also activates epithelial cells to release pro-inflammatory mediators, which amplify inflammation. Under smoke exposure conditions, epithelial cells and recruited inflammatory cells produce proteinases and oxidants that cause lung damage through alveolar septal cell apoptosis and destruction of the extracellular matrix (ECM).
Inflammation in Smoke Exposure
An increase in inflammatory cells has been documented in the lungs of patients with emphysema. Following smoke exposure, patients have an influx of macrophages and neutrophils into the lung. Neutrophils are implicated not only in disease initiation but also in exacerbations. As opposed to the human inflammatory response, the mouse experimental model of emphysema has a macrophage predominant inflammatory cell response, with groups demonstrating that mice lacking macrophages do not develop emphysema in smoke exposure conditions.
In addition to macrophages and neutrophils, B cells contribute to COPD development through the generation of lymphoid follicles within the parenchyma of patients with COPD. The size and number of these follicles is correlated with the severity of COPD.1 There is a possibly resultant auto-antibody production with anti-elastins, anti-epithelial, anti-tissue, and anti-nuclear antibodies all described in COPD.2,3 These auto-antibodies result in immune complex formation and complement mediated lung injury. Lung inflammation in COPD is perpetuated by a number of factors that may include latent adenoviral infections, which increase the expression of mediators and adhesion molecules,4 as well as recurrent bacterial and viral infections, which cause a chronic inflammatory state. Elastin and collagen fragments cause the release of matrikines that also amplify inflammation within the lung.5 All of these processes act together to result in the initiation and perpetuation of inflammation and resultant lung destruction (Figure 14,5,6).
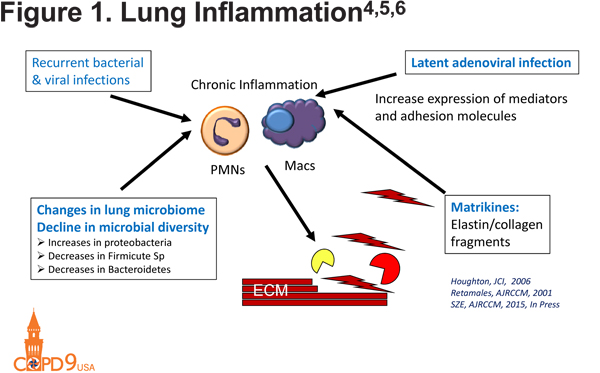
Proteinases in COPD
In the 1960s, an imbalance between proteinases and anti-proteinases was described in COPD. Main proteinase culprits include neutrophil serine proteinases, matrix metalloproteinases, and cysteine proteinases. Human data and transgenic mice have identified many involved proteinases in matrix destruction, including MMP-1, MMP-9, MMP-12, and neutrophil elastase.7-10 In addition to their direct effects on the ECM, proteinases also interact with other targets, augmenting inflammation or lung destruction. Examples include tryptases recruiting inflammatory cells into the lungs, neutrophil elastase inactivating tissue inhibitors of metalloproteinases, MMPs inactivating alpha-1 antitrypsin and macrophage inflammatory protein-1α. 10,11
Oxidative Stress in COPD
In addition to inflammatory and proteolytic consequences, smoke exposure has other detrimental effects on lung health. Cigarette smoke induces oxidative stress within the lung as a result of production of reactive oxygen species and reactive nitrogen species and a reduction of antioxidants including glutathione, vitamin A and E, superoxide dismutase and catalase (Figure 2). This oxidant-antioxidant imbalance is a result of reduced NRF2 activity. Oxidative stress has many downstream effects including inflammation, DNA damage and accelerated aging. Oxidative stress also induces epigenetic modifications including the inactivation of of histone deacetylase in continued patients with emphysema. The inactivation of HDAC2 results in continued pro-inflammatory gene expression and emphysema development.12
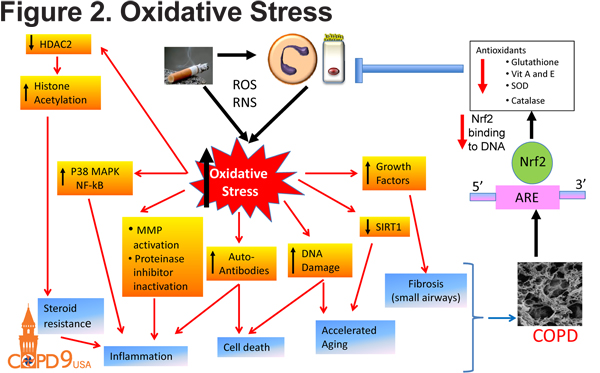
Alveolar Septal Cell Death in COPD
Finally, alveolar septal cell death results in emphysema development. Through programmed cell death (apoptosis), recycling of the cell components (autophagy) or cellular necrosis, there is a loss of alveolar cells and surface area resulting in emphysema.13-15 Apoptosis not only results in the phagocytosis of cell fragments, but also in the release of damage-associated molecular patterns that are pro-inflammatory. While well described in epithelial cells, the presence of these processes in the endothelium and leukocytes is not fully described.
In summary, lung injury in emphysema results from complex interactions between diverse processes including inflammation, proteinases, oxidative stress and cellular death. While well described in mice, key cells and culprits in human disease are not as clear. A better understanding of these processes in the human disease is essential to identifying opposing pathways to limit injury and progression.
Emphysema as a Disease of Deficient Tissue Repair/Maintenance
Cigarette smoke exposure causes stress responses within the lung, which can initiate processes critical to COPD development. The host must see cigarette smoke as a danger from the environment in order to initiate inflammatory and oxidative stress responses. The mammalian target of rapamycin (mTOR) is a sensor molecule that is critically important to the initiation of stress responses in the lung and alveolar maintenance. Following initiation, the disease progresses through a variety of factors. Cigarette smoke causes oxidative stress, which causes inflammation, apoptosis and autophagy, and protease-antiprotease imbalance, all resulting in the disruption of alveolar maintenance. Due to the unique nature of the lung interfacing directly with the environment, maintenance processes are believed to be ongoing, but in the setting of a stress such as cigarette smoke, critical alveolar maintenance programs are disrupted.
Vascular endothelial growth factor (VEGF), abundantly expressed in the lung, is required for the survival of endothelial cells, and as such was felt to be critical to the alveolar maintenance program. Inhibition of VEGF leads to airspace enlargement in an inflammation independent process.14 Inhibition of apoptosis or oxidative stress results in maintenance of the lung architecture.14 Rtp801, an inhibitor of mTOR, is activated in the VEGF inhibition model of airspace enlargement.16 In addition to smoke exposure conditions, Rtp801 is activated by stresses including radiation, chemotherapy, dexamethasone and other environmental triggers, linking Rtp801 to both initiation as well as disease progression.
In order to understand how COPD can be stopped and/or reversed, it is critical to understand the interactions between protease-antiprotease imbalance, apoptosis/autophagy and oxidative stress in tissue destruction17 These processes can be amplified by ceramides, endogenous mediators activated by cigarette smoke and VEGF receptor blockade, initiating pulmonary cell apoptosis, protease/antiprotease balance and oxidative stress,18 ultimately resulting in airspace enlargement. Activation of the ceramide pathway can engage destructive processes that can persist despite smoking cessation. Underscoring the possibility of airspace enlargement in the absence of inflammation, recent work in the Tuder laboratory has shown that endothelial cell death can create an endogenous wave of oxidative stress followed by elastolysis of the alveolar septa.
Disruption of normal lung maintenance leads to structural damage in emphysema. Telomere shortening leads to accelerated aging and disease exacerbations alter the lung microbiome. Each of these events results in inflammation and inflammasome activation. Telomere length in alveolar cells is also an important determinant of emphysema susceptibility.19
In summary, in order to initiate processes that lead to COPD development, cigarette smoke exposure must be partnered with other processes known to initiate emphysema development, including oxidative stresses, pro-apoptotic pathways and a protease rich environment. It is critical to understand these pathways as we attempt to understand disease susceptibility in target populations. COPD progression is another complex process, explaining why targeting of single pathways may be insufficient in halting the disease
Declaration of Interest
Dr. Stockley has served as an advisor for GlaxoSmithKline, CSL Behring, Chiesi, Boehringer Ingelheim and Grifols. He has served as a speaker for GlaxoSmithKline, CSL Behring, and Chiesi and has received grants from CSL Behring and Grifols. Dr. Owen owns shares in Pfizer, Bristol-Meyer Squibb, and Merck.