Running Head: COPD Pipeline
Funding support: Not applicable
Date of Acceptance: July 25, 2014
Abbreviations: inhaled corticosteroids, ICS; Food and Drug Administration, FDA; gold nanoparticles, AuNP; community-acquired pneumonia, CAP
Citation: Gross N. The COPD pipeline XXV. Chronic Obstr Pulm Dis. 2014; 1(2): 250-254. doi: http://doi.org/10.15326/jcopdf.1.2.2014.0143
Current Drug Development
It is appropriate to restart the COPD Pipeline column with a broad review of the subject(note: this COPD Pipeline appeared in another COPD-related journal over the past several years).
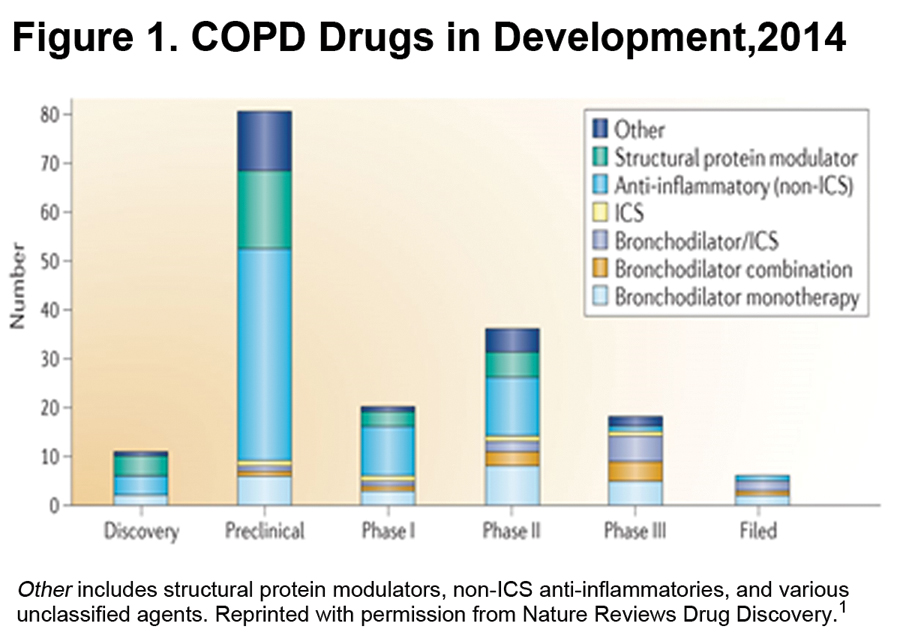
Figure 1 shows the overall COPD Pipeline by stage.1 Looking at drugs in late-stage development we see that the great majority are bronchodilator monotherapies, fixed combinations of bronchodilators, and fixed bronchodilator-inhaled corticosteroids (ICS) combinations. We have had many of these classes of agents for some years. The benefit of the new agents over previous ones is modest and is largely due to longer duration of action, convenience of combinations and improved delivery devices. Looking at agents in discovery and preclinical stages we see that there are still some bronchodilator monotherapies in early stages of development. One assumes that most if not all of them will, if successful, rapidly become components of fixed combinations as combinations become standard-of-care in COPD management.
The great majority of early stage agents are non-ICS anti-inflammatory agents (over 50 of them), and structural protein modulators (about 16 of them). The non-ICS anti-inflammatories include novel phosphodiesterase inhibitors, interleukin receptor antagonists, p38 MAPkinase inhibitors, calcium-activated potassium channel modulators, and possibly anti-oxidants among other agents. The structural protein modifiers target the airway remodeling process and mucus hypersecretion. Agents of those classes include elastase inhibitors, matrix-metalloprotease inhibitors, and possibly other protease inhibitors. We will report in more detail about these in future Pipeline columns. (Note: Smoking cessation drugs, of which there are at least 15 in various stages of development, are not included in the reference 1 report.)
It is very encouraging that the early drug development pipeline contains so many anti-inflammatory and protein modulator agents, and we eagerly anticipate their successful transition from the discovery and preclinical stages to late clinical trial stages and approval. It does not escape one’s notice however, that not a single member of such novel agents has made it to Phase III in the past, let alone approval. By contrast, agents of the ICS-bronchodilator sort are very unlikely to fail. The failure rate of non-ICS anti-inflammatories seems to have 2 elements. First, many agents in preclinical study stages do not make it to clinical stages. This is normal and entirely understandable. However, there is a second failure point at Phase II, where, for example, not a single non-ICS anti-inflammatory makes it through to the crucial Phase III. One can speculate on the reasons for this but one suspects that it is largely due to the absence of clear and acceptable end-points upon which pharmaceutical companies can be confident enough of their product to risk the enormous entailed investment in a pair of large, pivotal clinical trials.
Approval by the Food and Drug Administration (FDA) appears to be limited to objective criteria, e.g. a change in the rate of decline of lung function over time, or in the number of acute exacerbations. One doubts that these are the only relevant outcomes that concern patients. We need additional outcomes and one hopes that biomarkers will soon be reliable enough upon which to base an application for approval. Another important refinement will be phenotyping of COPD. The classification of treatments shown in Figure 1 takes no account at all of the fact that COPD is an extremely heterogeneous disorder. Thus the benefit of a new treatment that was appropriate for a particular patient phenotype could be obscured by its inclusion in a mixed patient population. Phenotyping may not be popular with parts of the pharmaceutical industry that would like their product(s) to be useful for the entire population of COPD patients but one senses that we have reached the one-size-does-NOT-fit all stage with our current treatments. For radical new advances, science and industry must look to treatments for the various, specific subtypes of COPD.
A COPD App?
Apps are being developed by startups that will cater to a variety of patient groups, among them patients with cystic fibrosis. Sixth Sense Healthcare Innovations2,3,4 has developed a spirometry test that uses an FDA-approved device coupled with a smartphone. The hope and aim is that indications of an acute exacerbation can be detected earlier and addressed before the condition worsens. Surely, with a little modification such an App could be developed for typical COPD patients.
Quadrivalent Influenza Vaccine
All COPD patients (and health care professionals) should receive the influenza vaccine each fall. The vaccine we have been using for years is trivalent, -protective against 2 influenza A viruses and 1 influenza B virus. The problem with the trivalent vaccine was that the experts had to choose between 2 very different B virus strains to include in the coming season’s vaccine. Thus, inadequate protection occurred when they made an incorrect choice of B strain. The quadrivalent vaccine, scheduled for release this fall, will protect against 2 influenza A viruses and 2 influenza B viruses, providing broader protection against influenza.
The quadrivalent vaccine is approved for all patients aged 3 years and older. There is alternatively a quadrivalent nasal spray vaccine approved for healthy people 2 through 49 years of age. Refer to the Center for Disease Control and Prevention’s table of 2014-15 approved influenza vaccines in the United States5 for more information. Influenza vaccine is not approved for children younger than 6 months of age, or for those who have had a severe allergic reaction to influenza vaccine.
Orphan Diseases and Drugs
There is a phenomenal gap between the cost of any commonly prescribed medication and the cost of an orphan drug. Why? The Orphan Drug Act6 was created in 1983 to incentivize the development of drugs for disorders that were so rare that pharmaceutical companies had little interest in developing treatments for them. For the purposes of the Act, a rare disease was defined as one that affects fewer than 200,000 individuals. (Europe and other countries have sometimes set the level differently). About 14,000 disorders have been designated rare by Orphanet, an organization that collects and categorizes rare diseases.7 Some of those diseases have been found in only a single individual. Before the Orphan Drug Act of 1983, a total of only 38 drugs had been approved for orphan disorders. Since 1983 the FDA has approved 353 orphan drugs. The market for orphan drugs is valued at $40 billion per year and growing rapidly,8 while the total market for pharmaceutical products was $329 billion in 2013. The majority of orphan drugs are being developed in the United States and more than 400 of them are currently in trials. Not surprisingly, the Orphan Drug Act has been called a great success. What is behind that success?
First, orphan drugs can command much higher reimbursements than another me-too bronchodilator or anti-hypertensive drug. Federal tax credits for orphan drugs can take 50% off research and development costs, on top of which sponsors are granted longer patent exclusivity. The FDA’s Fast Track and Breakthrough designations9 for orphan drugs make it a far less burdensome drive to approval and the Agency accepts that large, randomized double-blind controlled Phase III trials are unrealistic and often impossible. In addition, there are the facts that organ disease sufferers are easily identifiable, usually well motivated to enter a clinical trial, and often have the moral support and backing of an active patient advocacy group. Recruitment costs for a clinical trial of an orphan drug can be very modest. For all of these reasons, development of an orphan drug can be an attractive and potentially very profitable business plan.
Orphanet and the National Institutes of Health estimate that 5%-10% of rare diseases affect the lungs.10,11 About 40 are listed in the American Thoracic Society publication, Breathing in America.12 The designated orphan diseases in the COPD category include alpha-1 antitrypsin deficiency and cystic fibrosis (both of which affect more than 1 in 100,000 of the U.S. population), ciliary dyskinesia (between 1-10 per million), and elastin mutations of which there are only isolated reports.
Ataluren (formerly known as PTC124)
This is an interesting agent I have reported on previously.13 It is a small molecule that reportedly restores translation of an RNA message that contains an aberrant stop codon, a so-called protein-restoration therapy. It does this by making the ribosome less sensitive to premature stop codons (particularly ‘UGA’ codons) and is being studied for cystic fibrosis patients with a stop codon in the CFTR gene. This defect accounts for approximately 10% of cystic fibrosis cases worldwide, namely about 360 patients, but its frequency may be much higher in Ashkenazi Jews with cystic fibrosis. It results in a severe form of cystic fibrosis. A phase III randomized double-blind placebo-controlled trial of ataluren has recently been reported, 116 patients in each group.14 At 48 weeks, the primary endpoint, change in FEV1, was 3% better in the treatment group, a non-significant trend. The frequency of acute exacerbations, a secondary outcome, was 23% lower in the treatment group, also non-significant. Both outcomes, however, were greater and statistically significant in the subgroup of patients who were not receiving inhaled tobramycin. There were no significant safety concerns. Development will probably continue.
Aerosol Administration of Systemic Medications
Inhalation is becoming of interest as a route for the administration of agents that have usually been delivered parenterally or by some other route. The Journal of Aerosol Medicine and Pulmonary Drug Delivery devoted much of an issue in 2012 to the subject of administration of drugs and vaccines to the lungs by aerosol. The agents included some that were destined for a lung action such as chemotherapy, cyclosporine, bacteriophages for lung infections, treprostinil and surfactants. Among agents directed at non-pulmonary functions were insulin (Afrezza®, Mannkind), vaccines,15 interferon, heparin, salmon calcitonin, loxapine (Adasuve®, Alexza) for acute mania, and a wide variety of other agents encapsulated into PEG-liposomes.
Industry sees the pulmonary route as a promising area for drug delivery by inhalation. For this purpose, nanoparticulate drug delivery systems are being developed. One promising nano carrier is gold nanoparticles (AuNP). After their deposition in the lungs, inhaled AuNP’s come into contact with surfactant protein D which may facilitate both the systemic translocation of the carried agent as well as clearance. We may need to learn more about the benefits and problems associated with inhalation of agents and particles that are foreign to the lungs, particularly if use of the foreign inhalant is to be prolonged as for insulin, rather than one-time as for a vaccine.
Nemonoxacin
This novel antibiotic is C-8-methoxy nonfluorinated quinolone with enhanced in vitro activity against a wide variety of clinically-relevant pathogens, especially gram-positive bacteria, including multidrug-resistant Streptococcus pneumoniae and methicillin-resistant Staphylococcus aureus. It has a low propensity for engendering pathogen resistance due to the fact that bacteria become resistant to nemonoxacin only when 3 different mutations occur in their quinolone resistance-determining regions. Nemonoxacin shows greater efficacy than most of the widely used quinolones in the murine model of systemic, pulmonary, or ascending urinary tract infections. Nemonoxacin rapidly reaches maximum concentration in 1–2 hours after oral administration in the fasting state and has a relatively long elimination half-life similar to other fluoroquinolones. Approximately 60%–75% of the administered dose is excreted in unchanged form via kidneys over 24–72 hours. Phase II and III studies of oral nemonoxacin and Phase II studies of intravenous nemonoxacin have been completed in patients with community-acquired pneumonia (CAP), before which the Phase I studies of oral and intravenous nemonoxacin indicated sound tolerance and safety with healthy volunteers. In publications, an oral dose of either 500 mg or 750 mg nemonoxacin once daily for 7 days was as effective and as safe as levofloxacin 500 mg once daily for 7 days. Nemonoxacin was well-tolerated in patients with CAP, the most common adverse events following oral administration being gastrointestinal and nervous system adverse events similar to levofloxacin treatment. The Phase III studies of intravenous nemonoxacin for treating CAP and oral nemonoxacin for diabetic foot infection have not been published to my knowledge. The antibiotic is the subject of 7 trials in clinicaltrials.gov, 4 of them for CAP, and all but 1 are completed.16
Acknowledgements:
Information in the first section of this column was first published in Nature Reviews Drug Discovery by Nature Publishing Group, which provided permission to reproduce Figure 1. The author thanks Yasser Mushtaq, Senior Analyst of Global Business Intelligence Research and author of that report for use of the information and for his helpful suggestions. In addition, the author thanks Stanley Fiel, MD, of Morristown Memorial Hospital, Morristown, New Jersey for information about ataluren.
Declaration of Interest: none