Running Head: Clinical Trial Design for Alpha-1
Funding Support: Funding support for the Clinical Trial Design for Alpha-1 Antitrypsin Deficiency: A Model for Rare Diseases conference was provided by CSL Bearing, Grifols, the Alpha-1 Foundation, Alpha-Net and the Alpha-1 Project.
Date of Acceptance: February 17, 2015
Abbreviations: alpha-1 antitrypsin deficiency, AATD; alpha-1 antitrypsin, AAT; National Institutes of Health, NIH; National Institute of Neurological Disorders and Stroke, NINDS; NINDS-funded Network of Excellence in Neuroscience Clinicial Trials, NeuroNEXT; Antihypertensive and Lipid-Lowering to Prevent Heart Attack Trial, ALLHAT; Food and Drug Administration, FDA; institutional review board, IRB; Integrating Biology at the Bedside, i2b2; Patient-Centered Outcomes Research Institute, PCORI; Patient-Powered Research Networks, PPRN; National Heart, Lung and Blood Institute, NHLBI; Alpha International Registry, AIR; Alpha-1 Coded Testing, ACT; Genomic Research in Alpha-1 Antitrypsin Deficiency and Sarcoidosis, GRADS; Childhood Liver Disease Research and Education Network, ChiLDREN; Clinical Resource Center, CRC; antisense oligonucliotides, ASOs; forced expiratory volume in 1 second, FEV1;
Citation: Wanner A, Groft SC, Teagarden JR, et al. Clinical trial design for alpha-1 antitrypsin deficiency: A model for rare diseases. Chronic Obstr Pulm Dis. 2015; 2(2): 177-190. doi: http://doi.org/10.15326/jcopdf.2.2.2015.0132
Introduction
Historically, challenges to conducting clinical research in rare diseases have included fewer patient participants available for creating large cohorts; limited expertise for each rare disease and therefore, fewer researchers interested in conducting rare disease research; the lack of mass market profitability scenarios and limited potential for study sponsorship by industry partners; limited natural history information to utilize in designing treatment-oriented clinical trials; uncertainties about biomarkers and outcomes and, in general, a limited margin of error given the other challenges and the urgency to create new therapies for life-threatening illnesses.
However, potential solutions have been emerging over the past decade. An increased governmental focus on rare disease research1,2,3 and productive partnerships between patient advocacy groups and academic researchers, foundations, government regulatory scientists and industry have been formed creating new clinical research opportunities and optimism.4 Patient advocacy groups and disease-specific organizations have played a vital role in moving rare disease research forward as they recruit patients and assist in patient-researcher communications in multi-center trials.
These partnerships have created new challenges of addressing cultural, ethical, legal and social issues of data gathering and sharing across multiple populations, and have highlighted the necessity for the scientific community to embrace the need for innovation and flexibility in trial design, patient registries, bio-specimen repositories and natural history studies —all issues that create opportunities for greatly expanding rare disease research. In addition, perhaps the most critical current need in rare disease research is the development of appropriate biomarkers and surrogate and clinical endpoints for safety and efficacy in clinical trials. This challenge, along with the hurdles of small patient cohorts and the lack of natural history studies are the most pressing concerns facing rare disease research.
These challenges are clearly evident in a review of research in alpha-1 antitrypsin deficiency (AATD). This rare genetic disorder, characterized by chronic lung disease, liver disease or both, results from an abnormal folding of the alpha-1 antitrypsin(AAT) protein which is made primarily in the liver. The misfolded protein polymerizes and accumulates in liver cells and its secretion into the blood circulation is reduced. The polymerized AAT is cytotoxic and can lead to liver cirrhosis (gain of function) while the diminished level of circulating AAT, a serin antiprotease, exposes the lungs to unopposed protease activity resulting in lung tissue damage and chronic obstructive pulmonary disease (COPD) (loss of function). While intravenously administered AAT augmentation therapy exists for AATD lung disease, there is no specific treatment for AATD-related liver damage.
Research in both of these areas has faced the challenges of designing clinical trials with small patient populations and limited natural history studies while focusing on identifying appropriate biomarkers for use with safety and dose-ranging decisions in clinical trial designs and as signals of tissue damage.
In February 2014, the Alpha-1 Foundation convened a 2-day conference, Clinical Trial Design for Antitrypsin Deficiency: A Model for Rare Diseases, bringing together key stakeholders to discuss rare disease research challenges and how they have affected AATD research. This report is a summary of the discussions and conclusions from that conference.
Designing Trials with Small Patient Populations
The Need for Flexibility
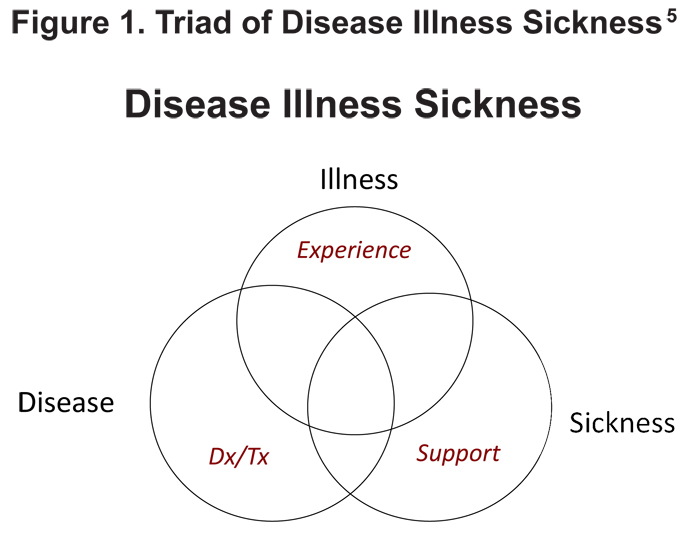
The traditional randomized controlled trial often is not a viable option in rare diseases, making flexibility in clinical trial design a necessity. The need for flexibility can be illustrated using Hoffman’s “Triad of Disease, Illness and Sickness” to define a health problem5 (Figure 1). Most clinical trials address the disease—its pathological process and to some extent the illness (the patient’s experience). However, a clinical trial must be flexible enough to address the entire triad which also encompasses sickness as it represents public and private societal responses (regulatory approvals, payers’ coverage policies, clinical adoption). This requirement is especially important in rare disease research.
Luce et al6 offer a matrix of evidence-based questions and activities to be asked and considered prior to a clinical trial (Figure 2). The questions, “Can it work?” (efficacy), “Does it work?” (effectiveness), and “Is it worth it?” (value to the patient and value to the payer) correspond to the 3 key evidence-based activities, evidence generation, evidence synthesis, and decision making. Within each of these questions and activities is the opportunity for flexibility. Designing a clinical trial that generates evidence on effectiveness and value to patients, providers and payers requires flexibility at the start of the process.

Many types of research models can be utilized for rare disease research (Figure 3). Selection of the type of study design for a particular clinical trial depends on 3 elements: 1) choice of the control used, 2) choice of the outcome and 3) nature of the disorder.The prudent choice of outcome measures in rare diseases can lead to answering study questions with fewer participants. However, caution must be executed to ensure that the selection of the study outcome is not based entirely upon the required sample size needed to conduct the study but is driven by the study question defined prior to the start of the trial.

Both continuous and longitudinal outcomes can be utilized with small sample sizes. Continuous outcomes describe those that are measured over an interval; they are qualitative outcomes and are distinguished from categorical outcomes which are nominal, ordinal or dichotomous. There is not a quantitative relationship between categories. Neither is particularly related or unrelated to time. Studies with continuous outcomes generally require a smaller sample size than those using categorical outcomes. Longitudinal outcomes measure the time until a response is observed, involve repeated observation of a few variables and ultimately requires the smallest sample size.
Studies can be designed to re-use the enrolled participants to increase study power when the number of study participants is limited. The often chronic or episodic nature of rare diseases lends itself to alternative study designs in that a type of stable background is often provided, allowing the disease process to return to baseline after an intervention is stopped. This allows for interventions to be tested in the same population a second time, as in crossover, factorial and N-of-1 designs.
The crossover design utilizes the same participant for different interventions and measures the response after each one. A factorial design asks 2 different study questions with participants used twice, answering both questions. N-of- 1 designs gather information on a participant several times, randomizing them continuously over multiple cycles of measurements of various interventions and controls/placebos. N-of-1 trials are considered to provide the strongest level of evidence about the existence of a causal relationship between a treatment and an outcome, and do not permit any generalization of the findings on the individual patient to any patient population, creating true personalized medicine. These trials may be combined through meta‐analysis or through a Bayesian random effects model and this combination provides a population estimate for treatment effectiveness while retaining the capacity to provide a distinct effectiveness estimate for each individual patient.
In enhanced trial designs wherepatients are used more than once, the trial itself determines which group to re-use. For example, in a randomized trial with an active intervention and a placebo, after the initial treatment, false responders to the placebo are re-randomized and offered treatment, allowing for intervention to be given to a new group while more accurately measuring the difference between the placebo group and the active group and providing a greater difference between responders and non-responders ultimately giving the study more power in the results.
Some flexibility may also be exercised when choosing the type of control utilized in a clinical trial. No controls may be considered for a disease with a devastating prognosis and would consist of establishing a cohort that is givenan intervention in the absence of any known, effective treatment. Historic controls may be used with a small or rare disease population in which the disease’s natural history is known and outcomes, given no treatment or standard of treatment, are also known. A new intervention would be compared to the patient’s historic experiences without the treatment. The clinical trial for glucarpidase, an orphan drug approved in 2012, is an example of a single-arm, open label trial using historic controls.7 Concurrent controls ensure that all aspects of the health care system, as much as possible, are held constant and can be used with both a randomized controlled study and with many alternative types of studies including a case control study. Self controls in which the patient is his or her own control allows for data to be collected from fewer patients at multiple time points.
Limitations: These alternative approaches do require a number of assumptions that may or may not be verifiable and those that can be tested for effect must be tested at the conclusion of the study, complicating the interpretation and reporting of the obtained data. The analyses of these alternative approaches are more complex than in traditional studies and require good statistical advice during the study’s design and development.
Adaptive Trial Design
As small clinical trials can only be powered to detect a large intervention effect, the importance of adequate study planning is magnified in them. Adaptive trial designs can address uncertainty about choices made during study planning by allowing for planned modifications to be built into the study design, based on analysis of the accumulating data.8 If initial assumptions are found to be incorrect, trial characteristics or design elements can be altered during the trial. However, these changes must be thoughtful and deliberate and not just the result of poor planning.
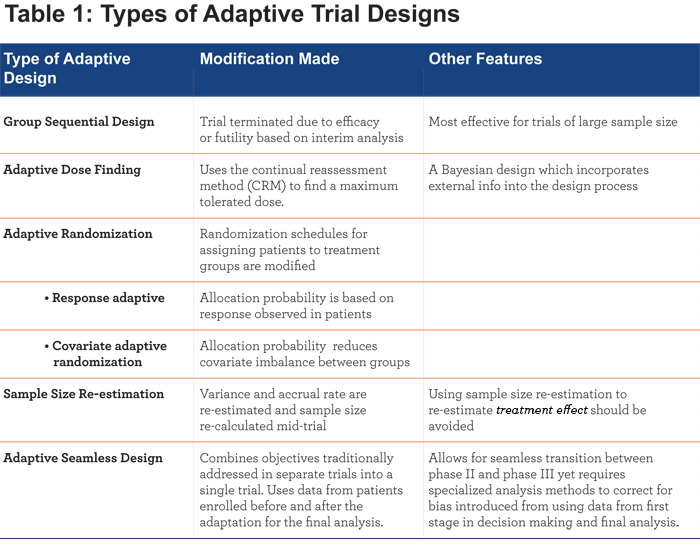
Adaptive designs often receive a great deal of attention as they are thought to be helpful in obtaining positive study results. However, the opposite is perhaps more often true. Specifically, adaptive designs can help accelerate a treatment development process by identifying ineffective treatments in a timelier manner than traditional clinical trial designs – thus allowing limited resources to be re-allocated to other potential treatments. There are numerous types of adaptations that have been proposed to date, including group sequential design, adaptive dose finding, adaptive randomization, sample size re-estimation, and seamless design.9 See Table 1.
The I-Spy 2 trial, which studied the use of additional chemotherapy drugs in breast cancer tumor reduction used an adaptive design that allowed the team to “learn as they went” using data from early patients to guide decisions about which treatments might be more useful for later patients, eliminating ineffective treatments and graduating effective treatments more quickly.10 Cost, time and the number of patients needed were all reduced while the pace of innovation was enhanced.
Limitations to Using Adaptive Designs: For diseases with a lengthy progression, such as AATD-related lung disease, there is less opportunity to see a response and adapt a trial in a timely manner to be effective. However, using a validated biomarker of the disease that correlates strongly in the short term to the long-term clinical outcome, could allow for the use of adaptive designs for these diseases. 9
Trials supported via traditional funding methods (National Institutes of Health [NIH], Veterans Affairs, voluntary health organizations) may be more challenged to use adaptive designs because these funding mechanisms may lack the flexibility to account for sample size modifications with re-estimations creating the need for supplementary funding. In addition, educating grant reviewers about an adaptive design’s unique statistical methods, which make it a valid study, may be required for it to proceed through peer review. 9
Adaptive trials generally require much more upfront planning as many scenarios must be addressed and simulations conducted to illustrate that the adaptation is successful across these scenarios. The time required for these simulations and trial justifications may offset any time saved by the adaptations. Burton et al11 stress the importance of creating detailed protocols for a simulation study along with defined, specific objectives and detailed procedures for data generation.
Industry has been more successful in using adaptive designs because it can utilize internal infrastructures designed for simulations and the corresponding code development. Budget limitations make the development of this type of infrastructure within academia unlikely, resulting in a growing divide between industry and academia in conducting adaptive trials.9 However, the National Institute of Neurological Disorders and Stroke (NINDS)-funded Network of Excellence in Neuroscience Clinical Trials (NeuroNEXT) is an example of a complex infrastructure being developed within an NIH-funded community which will allow for an increased ability to use novel trial designs including adaptive designs.12
Data management becomes critically important with adaptive designs. It must be monitored and recorded carefully as it will be used to make a mid-trial correction. In addition, analysis requires specialized methods to correct for bias introduced because the data from the first stage are used for both decision making and final analysis.
While appropriate statistical methods exist to support a much greater use of adaptive designs, most of the current research on adaptive designs is based on large sample theory, not small clinical trials. More research into the utilization of adaptive designs in small clinical trials is needed.
Pre-competitive Trial Design for Natural History Studies and More
As noted, lack of natural history studies, development of new biomarkers and studies on the molecular nature of the disorder are all challenging areas needing attention within the rare disease research community; yet generating this new knowledge is costly and not easily monetized. However, resources can be shared in a pre-competitive environment in which all partners participate equally and data are exchanged.
Pre-competitive research is a subset of translational research directed at advancing the methods and procedures needed for successful translation, and not on development of a specific product.13 It is often sponsored by groups to share the financial burden and allow involvement by government, academia, and the private sector, all of whom can benefit from the results.13
The Antihypertensive and Lipid-Lowering to Prevent Heart Attack Trial (ALLHAT) utilized a precompetitive approach with industry providing both financial support and study medications for this NIH-VA sponsored study involving 623 clinical centers. 14,15
The collaborations essential to pre-competitive research are focused on a common challenge that is needed for progress. This cannot be easily tackled by a single organization and cannot be exploited as an individual profit-making opportunity.16 Specifically, these types of collaborations must develop standards and methods that enable the necessary infrastructure to allow for effective data sharing and innovation and use these methods to generate, accumulate and integrate data.16
Therefore, new knowledge that is difficult and costly to generate, such as biomarker discoveries and disease model development, is more easily created by collaborators sharing in a precompetitive environment.
Increasing numbers of patient advocacy groups and foundations (the Multiple Myeloma Foundation, the Myelin Repair Foundation, and the Michael J. Fox Foundation for Parkinson’s Research) are utilizing a pre-competitive virtual pharma company format to expedite disease cures.16-19 Rather than funding several independent researchers on a one on one basis, these foundations are defining the research agenda, organizing research efforts and synthesizing knowledge. Through these collaborations, neglected and rare diseases with little market potential are able to advance and progress by driving collaboration and data sharing as a requirement for funding.16
In addition, by utilizing a virtual nature, funds are targeted to participants who have the greatest expertise and value. The foundations are able to provide project management and coordination across diverse research platforms, while pooling financial and human resources necessary to undertake large-scale projects. Potential problems include protecting intellectual property to make clinical trials and commercialization through industry profitable and encouraging large pharmaceutical companies to participate and perform the necessary trials.16
Through a precompetitive collaboration among 11 leading academic centers, major pharmaceutical companies, breast cancer patient advocates, the Food and Drug Administration (FDA), the National Cancer Institute, the Biomarker’s Consortium and the Foundation for the NIH, the I-SPY2 Trial process used shared data, tissue, tools, repositories, a common information management platform and used a sophisticated informatics portal to integrate and interpret complex and disparate data from many investigators.10
Biomarker/Endpoint Development
The FDA’s accelerated approval processcan provide some assistance with the challenging questions of addressing uncertainty about biomarkers or endpoints. Accelerated approval is given to new drug products that have been studied for safety and effectiveness in treating serious or life-threatening illnesses and which provide meaningful therapeutic benefit to patients over existing treatments. If uncertainty as to the relation of the surrogate endpoint to clinical benefit or of the observed clinical benefit to ultimate outcome exists, applicants for this approval are required to further study the drug, in a post marketing commitment, to verify and describe clinical benefit. Surrogate and intermediate clinical endpoints are considered in the accelerated approval process. A surrogate endpoint can be a subset of biomarkers such as a lab measurement, a radiographic image, or a physical sign or measurement thought to predict clinical benefit. An intermediate endpoint, while less common, is one that can be measured earlier than irreversible morbidity or mortality and is considered to likely predict a clinical benefit. 20-22
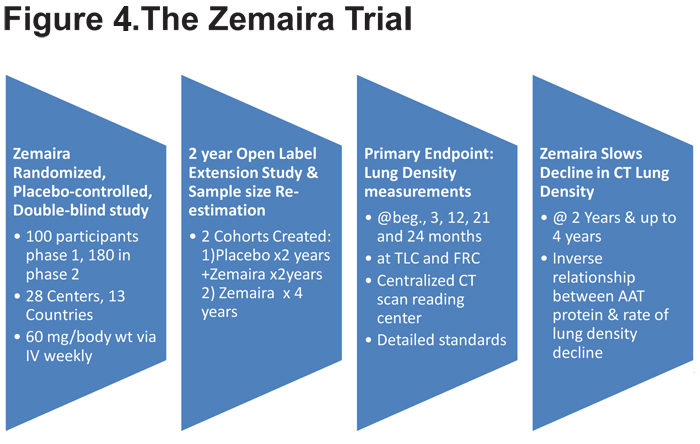
The approval of Zemaira for intravenous AAT augmentation therapy in 200323 provides an example of an FDA-stipulated post marketing commitment to conduct a clinical investigation, in 2 stages: Stage I was a pilot trial of clinically-meaningful endpoints and Stage II an adequately-powered study of clinically-meaningful endpoints contingent on the outcome and results of Stage 1. In this case, a single trial was used with a sample size re-estimation between phases I and II. In addition, a follow-up extension study allowed participants receiving placebo to switch to Zemaira for another 2 years.24 (Figure 4)
Regulatory Approach to Clinical Trial Designs
Drug development within the pharmaceutical industry has proven that the FDA is flexible on study design if there is sufficient evidence from investigators that the design preserves operating characteristics such as type 1 error and bias. While the statutory requirements for demonstrating effectiveness and safety are the same for rare and common diseases, and studies must demonstrate substantial evidence of clinical benefit, the statutory requirements do allow flexibility in how that demonstration is accomplished and how the standards are applied, given the wide range of uses for therapies.20-22 A recent review concluded that 60% of orphan drugs were approved without the need for 2 randomized controlled trials showing an effect, a typical requirement in drug trials.25 The FDA 2014 Guidelines provide further illumination of the agency’s stance on adaptive designs and which types of design it is more likely to accept.8
Patient Registries for Rare Disease Research Making data publicly available via a patient data registry provides the opportunity for multiple analyses and re-analysis of data which can lead to new results and conclusions. Over the last decade, many registries have been created. Most pharmaceutical companies have a registry for each drug they are developing. In addition, registries have been established to determine the clinical and cost effectiveness of tests/treatments, to measure safety or harm of products/treatments, to measure or improve quality of care, and assess the natural history of a disease. This includes estimating the magnitude of a problem, determining the underlying incidence or prevalence rate, examining trends of disease over time, conducting surveillance, assessing service delivery and identifying groups at high risk. Despite the proliferation of registries, there is still a deficit of data related to the natural history of most rare diseases, especially AATD-related lung and liver disease.
Currently, career disincentives, practice limitations, lack of funding and other barriers make building registries difficult.26 In addition, individual researchers must work with separate medical center information technology departments, and disparate institutional review board (IRB) approval processes that generate delays. Having a de-identified, publicly available, clinical research data repository seems to be a better option. In an ideal scenario, researchers could query clinical data absent of protected health information, and begin searching for the population that matches their research trial needs. Researchers would then apply for IRB approval after the population has been identified. Following IRB approval, the protected health information for the population could be accessed by the researchers to the extent and by the methods allowed by informed consent of the registry participants. Large database query tools such as Informatics for Integrating Biology at the Bedside (i2b2)27 are available in an open source environment to facilitate these newer approaches to registry science.
In addition, future registries/data networks will have an increased focus on patient engagement. The Patient-Centered Outcomes Research Institute (PCORI) describes the ideal data network as including patient involvement in the governance of the data use, having the capacity to collect patient-reported outcomes and covering large, diverse, defined populations from usual care settings. 28 PCORI, an independent non-profit organization authorized by Congress has a statute-mandated Rare Disease Advisory panel.29 It has established 2 types of data networks: Clinical Data Research Networks which must engage at least 2 health care systems and enroll at least 1 million patients and the Patient-Powered Research Networks (PPRN) which must address patients with a single condition, collect patient-reported data and involve patients in the governance.28
AATD-Specific Registries
Current patient databases within the alpha-1 community include: the National Heart Lung and Blood Institute (NHLBI) Registry (1988-1996), the Alpha-1 Foundation Research Registry, the Alpha International Registry (AIR), the Brigham and Women’s Sibling Pairs Database, the Alpha-1 Foundation DNA and Tissue Bank, the AlphaNet Database, the Alpha-1 Foundation contact database, databases unique to each pharmaceutical study, the Alpha-1 Coded Testing (ACT) study database, the Alpha-1 Foundation genetic counseling database and study specific databases (Genomic Research in Alpha-1 Antitrypsin Deficiency and Sarcoidosis [GRADS], and the Childhood Liver Disease Research and Education Network [ChiLDREN]).30-37 In addition, a Swedish National Neonatal Screening Program which screened 200,000 newborns for AATD in 1972-1974, has provided the largest unbiased AATD cohort information and is, after several decades, still following the population with future plans for additional studies that include imaging.38
The COPD Foundation was recently awarded a PCORI PPRN grant with plans to enroll 50-100,000 COPD patients in 18 months. The Alpha-1 Foundation is building a Clinical Resource Center(CRC) Registry, to eventually use its network of 82 Clinical Resource Centers across the United States with plans for shared data elements between the COPD PPRN clinical data warehouse and the Alpha-1 Foundation CRC registry. Using i2b2 programming, the Alpha-1 Foundation CRC Registry has the potential to make patient data available to interested alpha-1 research investigators as they plan clinical trials.
Alpha-1 Antitrypsin Deficiency Research: Challenges and Future Opportunities Alpha-1 antitrypsin deficiency research exemplifies all of the hurdles confronting rare disease research, with most of the recent AATD endeavors focused on identifying appropriate biomarkers and outcomes in AATD-related liver and lung disease in a small patient population with a slow-progressing disease.
AATD Liver Disease Research
Alpha-1 antitrypsin deficiency can cause liver disease in childhood and cirrhosis and/or hepatocellular carcinoma in adulthood. The important unresolved AATD liver disease research questions are:How is the liver damaged? Are there AATD specific markers of liver damage? What is the source of variability within AATD patients? (modifier genes vs. environment, or both?) How can it be treated? While some of these questions have been answered, appropriate non-invasive clinical endpoints for interventional studies are still under development. Quantifying the mutant Z protein accumulation in the liver via biopsy and quantifying the extent of fibrosis is a meaningful endpoint although it may not be considered a clinical outcome. Liver biopsy is invasive yet currently the most informative test. Elastography or magnetic resonance imaging may offer additional information through non-invasive means.39,40,41,42,43
Fibrosis or Biomarkers as Clinical Trial Outcomes
Fibrosis is measurable, fairly sensitive to change and relevant for the risk of developing cirrhosis, cancer and liver-related mortality, making it a potential primary or secondary endpoint for clinical trials, with the specific endpoint being lack of progression or reversal of fibrosis. In AATD, targeting mechanisms of liver injury is most likely to lead to improvement in fibrosis. Unfortunately no ideal, validated test of fibrosis quantification exists yet (liver biopsy is limited in its ability to assess fibrosis as it samples only 1/50,000 of the liver leading to sampling errors and is invasive, carrying a risk of morbidity and mortality), but newer imaging tests are promising.
Liver stiffness is a possible biomarker for fibrosis and can be measured by ultrasound-based transient elastography and magnetic resonance elastography.40,41,44 These imaging tests have a high accuracy rate for diagnosing advanced liver disease and a correlation with the clinical consequences of portal hypertension.
In addition, the fibrotest is widely used in Europe, in combination with the Fibroscan, to assess fibrosis in hepatitis C studies and provides a composite score of a simple grouping of alpha 2 macroglobulin, haptoglobin, apolipoprotein 1, total bilirubin, GGT and ALT measurements that correlate to a fibrosis stage.45,46 The combined use of fibroscan and fibrotest has allowed for, in large studies with long-term follow-up, a correlation to survival.47 However, more work is required in this area for these biomarkers to automatically have the same success in AATD-related liver fibrosis given the rareness of the disease and variability. Before fibrosis can be considered as a primary endpoint in clinical trials, a better understanding of the natural history of AATD-related liver disease in adults is needed.
Possible Therapeutic Approaches
With the goal of reducing the expression of the mutant protein, antisense oligonucleotides (ASOs) have been utilized to target the alpha-1 gene for treatment of AATD liver disease. ASOs can interact with both pre-mRNA in the nucleus and mature mRNA in the cytoplasm and can have multiple mechanisms of action including inhibition of the prime cap formation, inhibition of RNA splicing, activation of RNase H-mediated degradation or inhibition of polyadenylation.48 In addition, hepatocytes, where the accumulation of the mutant Z protein occurs, are very sensitive to ASO activity and several second generation ASO drugs have demonstrated efficacy across a broad range of patient populations, suggesting these could also be effective for alpha-1 patients. 49
To date, potent ASOs have been identified for human AAT in vitro.50 Guo et al have demonstrated that the administration of human alpha-1 ASOs in the PiZZ mouse model has stopped the disease progression after a 4 to 8-week treatment, reversed aggregate formation after a 20-week treatment, prevented aggregate formation in young mice and reduced the development of fibrosis in the PiZZ mice.
Additional approaches include gene silencing using RNA interference, small molecules that inhibit AAT polymerization, chaperones that promote intracellular AAT trafficking or autophagy enhancers.51
AATD Lung Disease
Existing and Future Therapeutic Approaches
A marketed therapy, intravenous AAT augmentation, is recommended for lung affected individuals who have AAT levels below 11 μM, documented evidence of airflow obstruction by pulmonary function tests51 and considered to have clinical benefit. 53,54
While AAT augmentation treatment has been an accepted treatment for AATD for over 20 years, it is not an ideal therapy because of its need for invasive IV access and life-long administration.
Aerosolized AAT therapy has been explored as a possible alternative to intravenous AAT augmentation given its ease of use and ability to deliver treatment directly to the airways and the lower respiratory tract, the desired target tissues in AATD-related lung disease. Most of the components needed to create aerosolized AAT for clinic use have been developed. These include the development of a highly purified form of AAT (examples: Aralast NP, Zemaira, Prolastin-C MP, Glassia), access to high efficiency deep lung delivery devices, the ability to determine the safety and appropriate dose of AAT, and the ability to show that the aerosolized AAT is reaching the interstitial space in the lung.
In the first studies of aerosolized AAT in humans, the number of neutrophils decreased in alpha-1 individuals receiving 8 weeks of aerosolized AAT.55,56 Those studies showed that at 4 and 8 weeks there is substantial increase in the amount of AAT appearing in the plasma indicating that it crossed from the epithelial side of the lung into the interstitium and finally the blood. Dry powdered aerosolized AAT, using recombinant AAT, has been shown, in a randomized, placebo-controlled study, to significantly increase the amount of AAT in the lower respiratory tract after daily dosage for 2 weeks but hypersensitivity can develop from impurities in the recombinant product.57 Robust outcome variables appropriate for rare disease studies and robust surrogate markers for a clinical trial phases I and II are still needed.
Biomarkers and Outcomes
Current clinically-meaningful outcome measures of AATD lung disease include mortality, morbidity, cognitive functioning and prevention of disease-accelerating events such as exacerbations. Specifically, measurement of forced expiratory volume in 1 second (FEV1), the rate, severity and duration of exacerbations and lung tissue destruction via CT densitometry are currently used as outcomes in AATD lung disease. Yet because these outcome measures have high variability, low sensitivity (FEV1) and depend on evolving methods (CT densitometry) they are not ideal for smaller/quicker studies, and surrogate endpoints are still needed for both proof of concept and even pivotal trials.
Phase II studies for AATD lung disease therapeutics focused on safety and dose ranging have had difficulty finding appropriate biomarkers with which dose ranging decisions can be made.
Current and potential AADT lung disease biomarkers include 1) molecular biomarkers (epigenetics,58 microbiomics, micro-RNAs), 2) functional biomarkers (6-minute walk test, BODE index), 3) measures of lung hyperinflation, 4) health status, 5) gene expression analyses, 6) markers of inflammation (interleukin-6, interleukin-8, leukotriene B4, tumor necrosis factor, fibrinogen, fibrinopeptides, polymorphonuclear leukocytes, C-reactive protein), and 7) markers of elastin degradation (desmosine/isodesmosine in bronchoalveolar lavage fluid, sputum, blood and urine).59-71 Some of these biomarkersmay not be specific enough to determine treatment effects in AATD.
Conclusions
Given the small patient populations available for rare disease research, flexibility must be exercised in clinical trial designs without compromising the validity and power of the trials. Several adaptive trial designs may offer opportunities for creating clinical trials with a smaller patient population, yet there are still limitations including potential funding concerns, a lack of understanding among protocol reviewers and the extensive infrastructure needed for data management and pre-planning simulations. Therefore, more dialogue among rare disease funders, regulatory agencies, statisticians and researchers is needed to address the current barriers to using adaptive designs.
Similarly, increased dialogue and partnerships among these stakeholders is needed to utilize the concept of pre-competitive trial design collaboration to address costly natural history studies, finding appropriate biomarkers and creating shared databases. In addition, while patient registries do exist within the rare disease community, more effort is needed to expand and intergrate these registries, and make them openly accessible to researchers for research queries while protecting patient privacy. Clearly, patient organizations must be engaged to accomplish this and local IRBs encouraged to embrace a new understanding of registries and their critical role in clinical trial development.
Taking AATD-related liver and lung disease as examples for the design of clinical trials in rare diseases, obtaining answers to critical questions could be facilitated by novel trial designs. Relative to liver disease, the key questions are: What is the rate of disease progression? What are the host and environmental factors that drive disease progression? What biomarkers correlate with liver disease presence, severity, and progression? A national liver disease registry that enrolls patients in a natural history study and identifies AATD patients with progressive disease for clinical trials is needed. Such a natural history study could identify patients who need drug therapy and when the drug therapy should be initiated. Currently there is no specific treatment for AATD-related liver disease and severely affected patients must resort to liver transplantation. However, new therapies are on the horizon and they will have to be clinically tested and face the challenges confronting rare disease trials.
In AATD-related lung disease research, finding clinically-relevant endpoints remains a critical focus. While the current AAT augmentation therapy provides some protection to lung tissues, it requires life-long repeated intravenous administrations. The promise of intrapulmonary augmentation therapy with aerosolized AAT is close to being realized; yet robust surrogate markers for phase I and II trials are still needed.
While much progress has been made in rare disease research over the past decade, many challenges still remain. Expanding and intensifying collaborative, innovative partnerships among researchers, patients, voluntary health organizations, industry, federal research grantors and regulators offers the most promise for future advancement. It was the purpose of the Clinical Trial Design for Alpha-1 Antitrypsin Deficiency: A Model for Rare Diseases conference to initiate this process.
Acknowledgements
The authors acknowledge that a freelance writer was engaged to provide assistance with the development of this manuscript. The Clinical Trial Design for Alpha-1 Antitrypsin Deficiency: A Model for Rare Disease conference was hosted by the Alpha-1 Foundation and chaired by Alpha-1 Foundation Scientific Director, Adam Wanner, MD, with Mark Brantly, MD; David Nelson, MD; Jean-Marc Quach, MBA; Robert Sandhaus, MD, PhD; Charlie Strange, MD, and Jeff Teckman, MD, serving as the conference’s steering committee. All listed authors served as conference speakers and contributed to the content and review of the final version of this manuscript.In addition, Kathryn O’Connell, MD, PhD, from the Rare Diseases Program, Office of New Drugs, Center for Drug Evalluation and Research, Food and Drug Administration, was also a speaker at the conference. Funding support for the conference was provided by CSL Bearing, Grifols, the Alpha-1 Foundation, Alpha-Net and the Alpha-1 Project.
Declaration of Interest
Dr. Brantley has received research funding from the Alpha-1 Foundatioin, Baxter Biologics, Grifols, Kamada Ltd, AGTC and CSL Behring. Dr. Edelman is an employee of CSL Behring Pharmaceuticals. Dr. McCaleb is an employee of Isis Pharmaceuticals. No other authors had any conflict of interests to declare.