Running Head: Lung Function in Alpha-1 Antitrypsin Deficiency
Funding Support: No funds were utilized to conduct this study
Date of Acceptance: July 13, 2015
Abbreviations: alpha-1 antitrypsin, AAT; orthotopic liver transplantation, OLT; pulmonary function tests, PFT; forced expiratory volume in 1 second, FEV1; AAT deficiency, AATD; forced vital capacity, FVC; single lung transplantation, SLTx; bronchiolitis obliterans syndrome, BOS; percent predicted, % predicted; Global initiative for chronic Obstructive Lung Disease, GOLD
Citation:Tejwani V, Wang XF, Stoller JK. The natural history of lung function in severe deficiency of alpha-1 antitrypsin following orthotopic liver transplantation: A case report. Chronic Obstr Pulm Dis. 2015; 2(4): 290-295. doi: http://doi.org/10.15326/jcopdf.2.4.2015.0141
Introduction
PI*ZZ alpha-1 antitrypsin (AAT) deficiency poses risk for both lung and liver disease, with different pathogenetic mechanisms for each. The associated emphysema is caused by a toxic loss of function in which deficient serum and lung AAT levels cause a depleted proteolytic screen,1 posing risk for unchecked elastolysis in the lung and resultant emphysema. Through separate mechanisms, the proinflammatory effect of Z polymers that are either produced in the lung by alveolar macrophages or secreted from the liver and perfuse the lung, putatively recruit neutrophils to the lung, thereby increasing the proteolytic burden and promoting emphysema.2-5 The relative contributions of each of these mechanisms to the emphysema associated with PI*ZZ AAT deficiency (AATD) is unknown.
In contrast to the pathogenesis of lung disease in AATD, liver disease in PI*ZZ AATD results from a toxic gain of function in which unsecreted Z polymers that are trapped in the hepatocyte confer a risk for fibrosis, cirrhosis, and hepatocellular carcinoma.1 For PI*ZZ individuals with end-stage liver disease, orthotopic liver transplantation (OLT) with a normal liver graft represents the only currently available therapy, with the prospect that the normal (PI*MM) donor liver will produce normal serum levels of AAT, thereby obviating subsequent lung risk. While OLT theoretically averts the subsequent risk of emphysema, the natural history of lung function following OLT for PI*ZZ AAT deficiency has been sparsely studied, with only 2 series describing a total of 24 patients.6,7 To extend understanding of the natural history of lung function following OLT in PI*ZZ AATD, the current report describes a patient with PI*ZZ AATD who underwent OLT 6 years after single lung transplantation, the latter prompting multiple pre- and post-OLT spirometry measurements and uniquely allowing a comparison of the rates of change of FEV1 and FEV1/ forced vital capacity (FVC) before versus after OLT.
Case Report
A 74 year-old female underwent both single lung transplantation (SLTx) for emphysema and later OLT for cirrhosis, both due to effects of PI*ZZ AATD. The SLTx was performed at the age of 59 (on April 23, 2000) and the OLT was performed 6 years later (on June 21, 2006). She had smoked ≤ 1 pack/day for 10 years (from age 40 to age 50). The diagnosis of PI*ZZ AATD was first established at age 55. Despite treatment with oxygen and with intravenous augmentation therapy8 for 2 years, she experienced progressively debilitating dyspnea. Evaluation for lung transplantation was initiated in 1998 and she received a right SLTx on April 23, 2000. She received neither regular blood transfusions nor augmentation therapy following lung transplantation. Surveillance bronchoscopy thereafter showed no evidence of acute cellular rejection as late as 19 months post-SLTxt (to November 13, 2001).
Thrombocytopenia and elevated hepatocellular enzymes were first observed in late 2004 and prompted an abdominal ultrasound, which showed ascites and splenomegaly. In September 2005, cirrhosis was confirmed by liver biopsy, which also showed the presence of periodic acid Schiff positive-diastase-resistant globules. Her liver course was complicated by re-accumulating ascites, repeated paracenteses, hepatic hydrothorax, and an episode of hepatic encephalopathy, culminating in liver transplantation on June 21, 2006. Early bronchiolitis obliterans syndrome (BOS) was first noted in her medical record on October 7, 2009, 9 years post-SLTx.
Spirometry was performed frequently as part of her routine post-SLTx surveillance, with a total of 50 and 22 pulmonary function test (PFT) sessions performed pre- and post-OLT respectively.
Methods
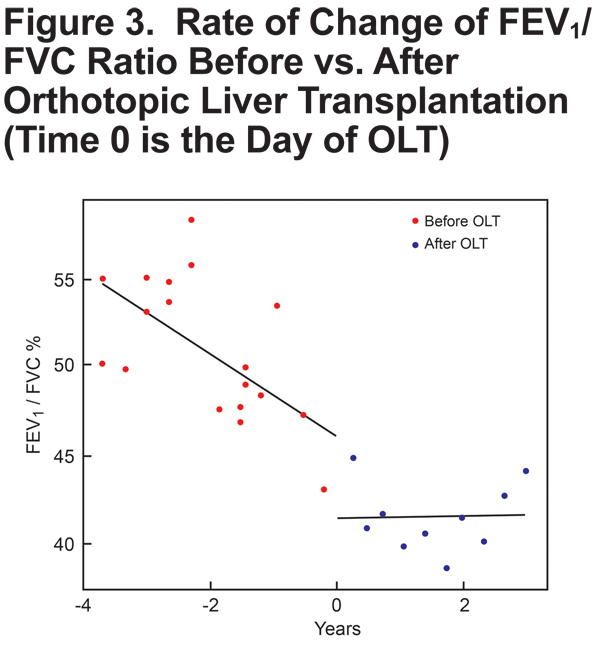
A segmented regression approach, in which the independent variable is partitioned into intervals and a separate line segment is fitted to each interval, was utilized. These separate line segments (i.e., before OLT versus after OLT) were compared to assess whether the rate of lung function before OLT differed from that after OLT. Rates of change of FEV1, FEV1 percent predicted (% predicted), and of FEV1/FVC over time (called slopes) were calculated by fitting values performed pre-OLT and then again post-OLT with separate lines. All statistical tests are two-tailed and values of p<0.05 are considered significant. Statistical analyses were performed using SAS 9.3 software (SAS Institute, Cary, NC).
To eliminate the confounding effects of surgery and intercurrent clinical events on lung function measurements, spirometry sessions were censored from inclusion for several reasons, i.e., when spirometry was:
- Performed within 2 months following OLT or SLTx,
- Performed during clinic visits for intercurrent respiratory events, like upper respiratory tract infections, or
- Performed after the reported onset of BOS.
Also, only PFTs performed while she was under active surveillance or undergoing clinical follow-up at Cleveland Clinic were used in the analysis. Altogether, after censoring, 19 sets of PFTs pre-OLT and 10 sets of PFTs post-OLT were used to calculate FEV1 and FEV1/FVC slopes.
Results
Following OLT, the patient's serum AAT level normalized (170 mg/dL, normal range 100-220 mg/dl), consistent with her having received a normal (PI*MM) donor liver.
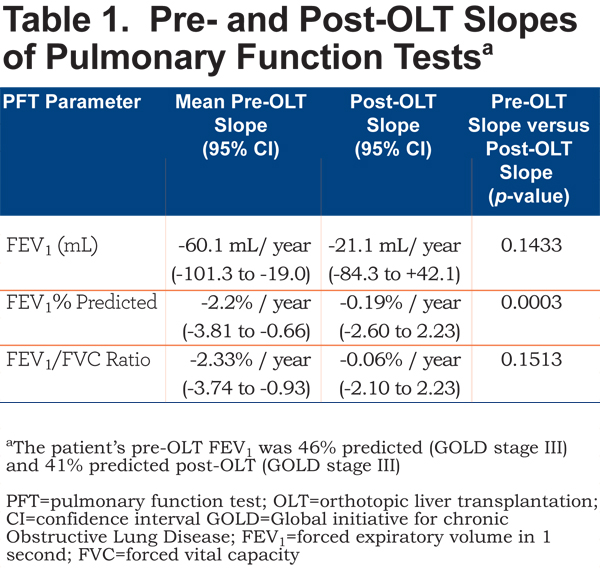
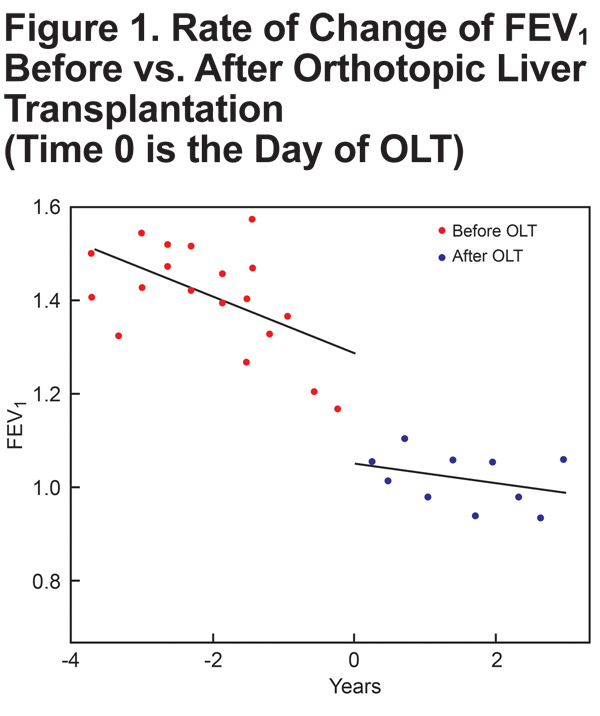
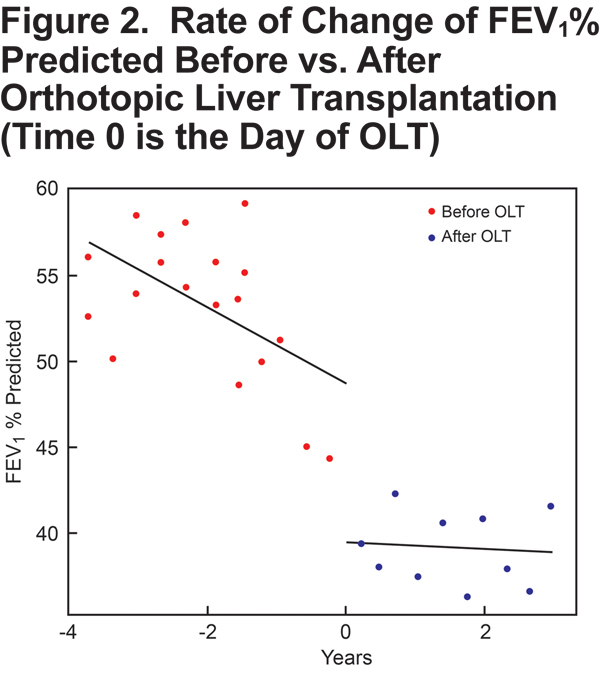
Slopes for all PFTs (FEV1, FEV1 % predicted, FEV1/FVC) trended to less negative post-OLT than pre-OLT (Table 1, Figures 1-3), suggesting a slower rate of lung function decline following OLT. For example, before OLT, her FEV1 declined at a rate of 60.1 ml/year, whereas post-OLT, the rate of decline decreased to 21.1 ml/year (p = 0.143). The slope of FEV1% predicted slowed significantly after OLT (p = 0.003).
Discussion
The main observation from this single patient's 130-month course surrounding an OLT for PI*ZZ AATD-associated cirrhosis is that the rates of decline of FEV1, FEV1 % predicted, and FEV1/FVC numerically slowed post-OLT compared to pre-OLT. Liver transplantation was associated with restoration of the normal serum AAT level and of the rate of lung function decline to that of normal age-related decline (~20 ml/year).9 This normalization of the post-OLT FEV1 slope in a patient with a single remaining native lung may shed light on the relative contributions of the 2 mechanisms of emphysema, i.e., unchecked proteolytic screen due to low AAT levels versus the proinflammatory effect related to chemotaxis by Z polymers produced in alveolar macrophages and secreted from the liver.5 Specifically, to the extent that the rate of FEV1 decline normalized post-OLT, this patient’s course suggests that restoring the normal serum AAT levels exerted a greater effect on preventing emphysema progression than local4 or hepatic Z polymer production5 contributed to furthering emphysema.
This observation extends current understanding of the natural history of lung function following OLT for PI*ZZ AAT deficiency by offering, to our knowledge, the first assessment of lung function that is based on multiple pre- and post-OLT spirometry measurements, thereby allowing calculation of rates of FEV1 change. In the 2 prior reports,6,7 the assessment of lung function change before versus after OLT was based on comparing only single pre-OLT with single post-OLT lung function measurements, with the exception of a single patient for whom 4 post-OLT spirometry measurements were available. In the first series of 7 patients by Jain et al,6 pre-OLT PFTs were performed an average of 5.6±3.4 months before OLT and demonstrated a mean FEV1 of 2.6±0.9L compared to a mean post-OLT FEV1 of 2.7±1.2L performed 30.3±18 months after OLT. A single patient had 4 sets of PFTs performed from 12-59 months post-OLT, which demonstrated a stable FEV1. In a more recent report by Carey et al,7 PFTs were performed an average of 8.8±12.9 months pre-OLT for 40 PI*ZZ patients; the mean FEV1 pre-OLT was 2.79±1.10L and, in the 17 patients with available measurements, the mean post-OLT FEV1 (performed a mean of 49.5±36.8 months post-OLT) was 2.73±1.34L. Neither the change in FEV1 nor FVC achieved statistical significance, though the FEV1/FVC ratio was significantly lower post-OLT than pre-OLT.
In interpreting PFT changes from pre- to post-OLT, documenting that post-OLT serum AAT levels normalized is important. Cases have been reported in which donor livers to AATD recipients were inadvertently also from AAT deficient donors,10,11 leaving open the possibility that accelerated lung function decline post-OLT reflects ongoing deficiency of AAT. As in our patient, post-OLT AAT serum levels were also measured and shown to be normal in a few of the 17 patients reported by Carey et al.7
Though this analysis is the first, to our knowledge, to analyze multiple pre- and post-OLT measurements in a single PI*ZZ individual, several shortcomings of this report warrant comment. First and most obviously, information from a single patient experience must be viewed with caution regarding robustness and generalizability. Also, confidence intervals around the slope estimates in this report remained wide despite the availability of multiple measurements, underscoring the shortcoming of prior estimates that are based only on single pre- and post-OLT measurements. Clearly, more robust assessment would require even more pre- and post-OLT measurements in multiple PI*ZZ patients. A second limitation is that emphysema progression was assessed only by FEV1 change rather than by CT densitometric assessment, which has been shown to detect emphysema that is unapparent on PFTs.12 Accordingly, it is possible that our observation of a normalized rate of FEV1 decline post-OLT under-estimates continued emphysema progression. Third, OLT in our patient was associated with an unexplained step drop-off of FEV1 by ~200 ml (Figure 1). Dawkins et al13 showed that rates of FEV1 decline are lower in AAT deficient individuals with higher Global initiative for chronic Obstrucitve Lung Disease (GOLD) strata than in those with better preserved FEV1 values, with the greatest slowing of mean FEV1 decline in the transition from GOLD stage III to GOLD stage IV. While this observation raises the possibility that the slowing of our patient’s FEV1 slope after liver transplantation relates to the floor effect of having a lower FEV1 post-OLT, the ~200 ml FEV1 drop post-OLT was unassociated with a change in her classification as GOLD III.
Finally, our findings lessen the likelihood that local pulmonary Z polymer production primarily drives emphysema progression but do not separate the 2 contributions of transplanting a normal liver, i.e., restoring normal serum AAT levels and eliminating liver-derived Z polymers. Tan et al5 reported a single patient in whom circulating Z AAT polymers were detected pre-OLT and decreased substantially following OLT. The possibility that eliminating circulating AAT polymers was responsible for the slowing of lung function decline could only be excluded by both lavage and serum analysis of Z AAT polymers before and after liver transplantation. As such tests were not performed as part of routine clinical care, this mechanism of emphysema progression cannot be completely discounted. Still, the observation that circulating Z polymers lessen post-OLT is also based on only a single patient5 and, although circulating Z polymers decreased post-OLT in that patient, they did not disappear completely. Also, attributing the slowing of emphysema progression solely to eliminating circulating Z polymers post-OLT overlooks the substantial evidence that augmentation therapy slows emphysema progression.2,14-16 As such, this patient’s experience suggests that the slowing of emphysema progression post-OLT likely relates to restoring AAT levels either alone or in concert with eliminating liver-derived circulating Z polymers.
Declaration of Interest:
Vickram Tejwani has no conflicts of interest to declare. Xiao-Feng Wang has no conflicts of interest to declare. James K. Stoller has served as a consultant to Grifols, Baxter, Kamada, Arrowhead Research, and CSL Behring and serves on the Board of Directors of the Alpha-1 Foundation.