Running Head: Efficacy and Safety of Glycopyrrolate
Funding support: The study was supported and funded by Novartis Pharma AG, Basel. Writing support was funded by Novartis Pharma AG, Basel, Switzerland.
Date of Acceptance: December 9, 2015
Abbreviations: Glycopyrrolate Effect on syMptoms and lung function study, GEM2; twice daily, b.i.d.; forced expiratory volume in 1 second, FEV1; area under the curve, AUC; St. George’s Respiratory Questionaire, SGRQ; Transition Dyspnea Index, TDI; Global initiative for chronic Lung Disease, GOLD; long-acting muscarinic antagonist, LAMA; long-acting β2-agonist, LABA; inhaled corticosteroid, ICS; chronic obstructive pulmonary disease, COPD; U.S. Food and Drug Administration, FDA; forced vital capacity, FVC; modified Medical Research Council, mMRC; electronic diary, e-diary; minimal clinically important difference, MCID; COPD Assessment Test, CAT; adverse event, AE; major adverse cardiovascular events, MACE; cardio-and cerebrovascular, CCV; full analysis set, FAS; per protocol set, PPS; Baseline Dyspnea Index, BDI; glycopyrrolate, GLY; placebo, PBO; confidence interval, CI; odds ratio, OR; serious adverse effects, SAEs; Friederica’s corrected QT interval, QTcF; least squares means, LSM
Citation: Kerwin E, Siler TM, Korenblat P, et al. Efficacy and safety of twice-daily glycopyrrolate versus placebo in patients with COPD: the GEM2 study. Chronic Obstr Pulm Dis. 2016; 3(2): 549-559. doi: http://doi.org/10.15326/jcopdf.3.2.2015.0157
Introduction
The current Global initiative for chronic Obstructive Lung Disease (GOLD) strategy1 and American Thoracic Society/European Respiratory Society guidelines2 recommend inhaled bronchodilators, such as a long-acting muscarinic antagonist (LAMA) alone or in combination with a long-acting β2-agonist (LABA) or inhaled corticosteroid (ICS), as the mainstay for treatment of patients with chronic obstructive pulmonary disease (COPD). Glycopyrrolate, also known as glycopyrronium bromide, is a fast onset (statistically significant bronchodilation within 5 and 15 minutes post-dose) and long-acting (sustained 24-hour bronchodilation) muscarinic antagonist (LAMA), developed for maintenance treatment in patients with COPD.3-5 Preclinical studies with glycopyrrolate have demonstrated high affinity and slow dissociation from muscarinic receptors which supports the prolonged bronchodilation effect in patients with COPD.6 Various global clinical studies have demonstrated that treatment with glycopyrrolate 63 µg (equivalent to glycopyrronium 50 µg) administered once-daily offers substantial benefit to patients with moderate to severe COPD with improved lung function, dyspnea, health status, rescue medication use, with overall good safety profile 4,5,7 and is an approved treatment in more than 80 countries including the European Union, Brazil, Japan, Canada, Switzerland and Australia, excluding the United States. In the United States, a separate phase III clinical trial program with glycopyrrolate 15.6 µg (equivalent to glycopyrronium 12.5 µg) was developed that included 2 identical pivotal 12-week efficacy and safety studies, Glycopyrrolate Effect on syMptoms and lung function study (GEM1) and GEM2 and a long-term safety study, GEM3. The dose selection was based on a dose-ranging study8 showing statistically significant and clinically relevant improvements in trough forced expiratory volume in 1 second (FEV1) with twice-daily glycopyrrolate 15.6 µg treatment and on discussions with the U.S. Food and Drug Administration (FDA). Seebri® NeohalerTM (glycopyrrolate) 15.6 µg is now approved in the United States for the long-term maintenance treatment of airflow obstruction in patients with COPD.
This manuscript presents the results of the 12-week Phase III GEM2 study that assessed the efficacy and safety of glycopyrrolate 15.6 μg b.i.d. compared with placebo in COPD patients with moderate-to-severe airflow limitation (GOLD 2 and 3 according to the GOLD 2011 strategy).
Methods
Study Design and Treatment
GEM2 was a 12-week multicenter, randomized, double-blind, parallel-group, placebo controlled study (ClinicalTrials.gov registration number: NCT01715298). The study comprised an initial wash out period (duration of 7 to 1 days depending on washout required for prior medications), a run-in period of 2 weeks, followed by a 12-week randomized treatment period, and a 30-day safety follow-up period. Patients were randomized after the run-in period to receive glycopyrrolate 15.6 µg b.i.d. or placebo (both delivered via the single-dose dry-powder inhaler [the NeohalerTM device; Novartis, Basel, Switzerland]) for the following 12 weeks using an allocation ratio of 1:1 (Figure 1).
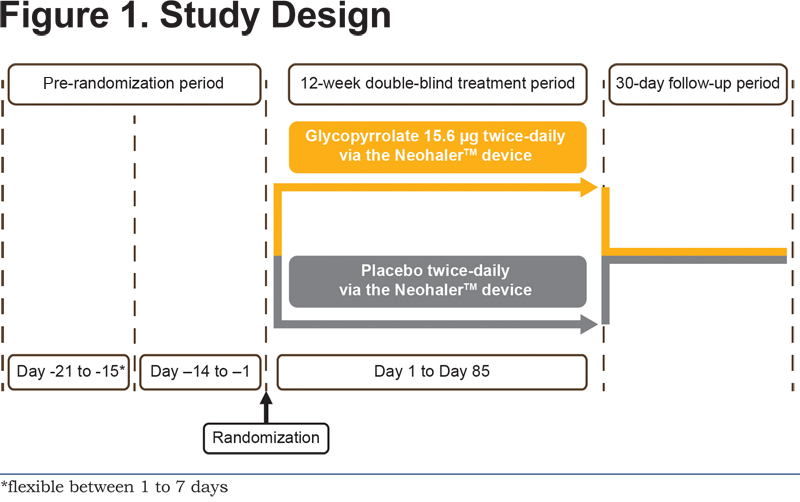
Additional details of the randomization and blinding procedures are included in the online supplementary material. This study was conducted at 64 centers within the United States. The first patient was enrolled on November 26, 2012 and the last patient visit was completed on December 26, 2013. Albuterol (100 µg/puff administered from the pressurized metered-dose inhaler) was allowed as a rescue medication. The continuation of inhaled corticosteroid (ICS) monotherapy at a stable dose regimen was permitted as COPD background therapy. This study was submitted to the FDA, was approved by the institutional review boards/independent ethics committees/research ethics boards of participating centers, and was conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonization Good Clinical Practice guidelines. All patients provided written informed consent before participating in the study.
Patients
The study population included male and female patients aged 40 years or older with moderate-to-severe airflow limitation (GOLD 2 or 3 as defined in GOLD 2011), current or ex-smokers with a smoking history of at least 10 pack years, post-bronchodilator FEV1 ≥30% and ˂80% of predicted value, post bronchodilator FEV1/forced vital capacity (FVC) ratio ˂0.70, and a modified Medical Research Council (mMRC) Dyspnea Scale grade of at least 2 at the run-in visit. Key exclusion criteria included history of asthma and COPD exacerbation requiring treatment with antibiotics, systemic corticosteroids (oral or intravenous), and/or a hospitalization within 6 weeks before screening or during screening and run-in. Detailed inclusion and exclusion criteria are provided in the online supplementary material.
Study Objectives
The primary objective was to demonstrate the superiority of glycopyrrolate 15.6 µg b.i.d compared with placebo for FEV1 standardized area under curve (AUC) between 0 and 12 hours post dose (FEV1 AUC0-12h) at Week 12. The secondary objectives were evaluation of glycopyrrolate compared with placebo in terms of lung function through trough FEV1 (mean of FEV1 measured at 23:15 h and 23:45 h post previous morning dose) at Day 2 and Week 12, standardized FEV1 AUC between 0 and 4 hours post-dose (AUC0-4h), 4 to 8 hours post-dose (AUC4-8h), and 8 to 12 hours post-dose (AUC8-12h) on Day 1 and Week 12, FEV1 and FVC recording at the following time points relative to the morning dose: 5 and 15 min, 1, 2, 4, 8, and 12-hours post dose on Day 1 and Week 12. Additional lung function efficacy objectives were peak FEV1 and FVC during 4 hours post morning dose on Day 1 and Week 12 and trough FVC on Day 2 and Week 12. The time (min) to achieve ≥100 mL improvement in FEV1 from baseline on Day 1 was also evaluated to determine the onset of action.
Other secondary objectives included evaluation of dyspnea (assessed via the Transition Dyspnea Index [TDI] focal scores), health status (assessed via the St. George’s Respiratory Questionnaire [SGRQ] total score) at Week 12, rescue medication use, and COPD symptoms reported using the patient electronic diary (e-diary) over the 12 week treatment period, which also confirmed study compliance. The percentage of patients who achieved the minimal clinically important difference (MCID) in SGRQ total score and TDI focal score in the glycopyrrolate and placebo treated groups was also analyzed. An exploratory objective of the study was to evaluate health status using the COPD assessment test (CAT) at Week 12.
Safety assessments included treatment-emergent adverse events (AEs), monitoring of vital signs (pulse rate, systolic and diastolic blood pressure), electrocardiography, and laboratory analyses (hematology, clinical chemistry assessments, and urinalysis). All serious cardio- and cerebro-vascular events (CCV), atrial fibrillation and atrial flutter, and all cases of death that occurred between randomization and the end of the follow-up period were evaluated by an independent adjudication committee. Serious CCV events were adjudicated by major adverse cardiovascular event (MACE) outcome and atrial fibrillation/flutter events were adjudicated based on the new onset or recurrence/persistence of events.
Statistical Analysis
The full analysis set (FAS) included all randomized patients who received at least 1 dose of the study drug. The per-protocol set (PPS) included all patients in the FAS who did not have any major protocol deviations. The safety set included all patients who received at least 1 dose of the study drug. The FAS was used for the analysis of the primary objective and all other efficacy variables. The PPS was used for the supportive analysis of the primary variable. The safety set was used in the analysis of all safety variables.
FEV1AUC0-12h was calculated using the trapezoidal rule divided by length of time (12 hours).9 For the analysis of the primary efficacy endpoint (FEV1 AUC0–12h), a mixed model for repeated measures was used. The model contained terms for treatment, baseline FEV1, baseline smoking status, baseline ICS use, visit, treatment x visit interaction and baseline FEV1 x visit interaction, with an unstructured covariance matrix. Secondary endpoints were analyzed using the same mixed model as used for the primary endpoint, with the respective baseline values replacing baseline FEV1 as a covariate as necessary. The proportion of patients who achieved the MCID in the SGRQ total score and TDI focal score was analyzed using a logistic regression model. Details on the sample size estimation are given in the online supplementary material.
Results
Patient Disposition and Baseline Characteristics
Of the 1144 patients screened, 432 were randomized to glycopyrrolate (n=216) or placebo (n=216), and 414 patients (96%) completed the 12-week planned treatment phase (glycopyrrolate, n=209 [96.8%]; placebo, n=205 [94.9%]). Patients could continue to participate in the 12-week planned treatment phase even if they had permanently discontinued the study medication. The most common reason for study discontinuations was patient decision (glycopyrrolate, n=7; placebo, n=9) and 2 patients were discontinued in the placebo group due to lost to follow up.
Patient demographics and baseline clinical characteristics were comparable between the treatment groups (Table 1). Most of the enrolled patients were men (58.8%) and white(86.8%) with moderate airflow limitation (61.1%). Most of the patients belonged to the GOLD 2011 Group B (57.6%) and did not have COPD exacerbations in the year prior to study entry (78.0%).
Efficacy
Lung Function
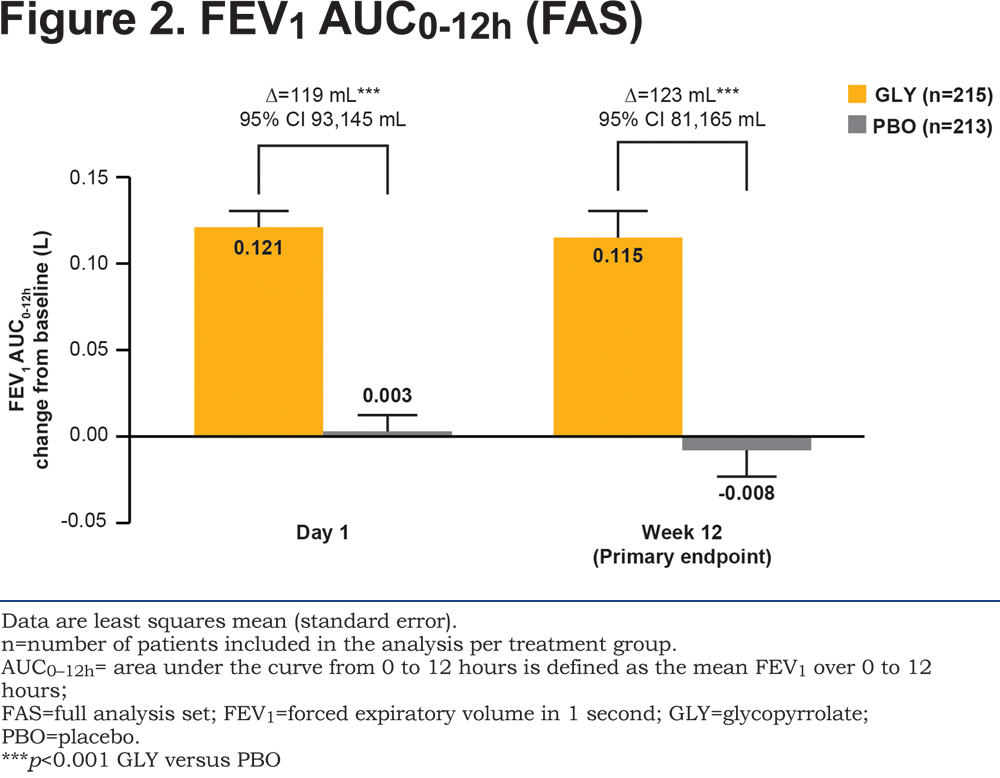
The study met its primary objective with glycopyrrolate demonstrating significant improvement compared with placebo (p<0.001) in change from baseline in FEV1 AUC0-12h at Week 12 (Figure 2). Both at Day 1 and Week 12, FEV1 AUC0-12h was higher with glycopyrrolate (p<0.001) compared with placebo, with a statistically significant and clinically meaningful treatment difference10 of 119 mL and 123 mL, respectively (Figure 2). The results of the PPS analysis (Week 12) were consistent with the primary analysis (treatment difference 128 mL; 95% confidence interval 84, 171 mL; p<0.001).
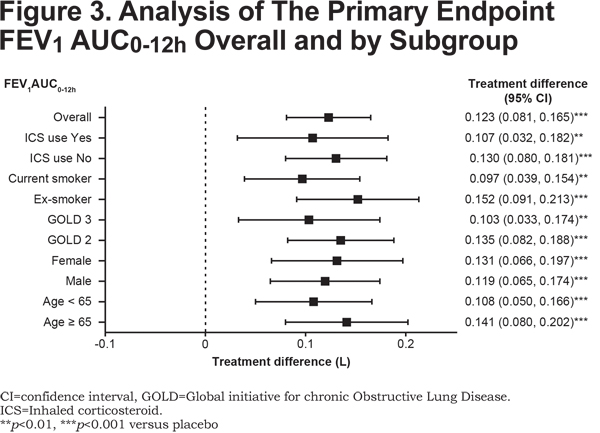
Improvement in FEV1 AUC0-12h with glycopyrrolate versus placebo in all the subgroups based on age, sex, airflow limitation, baseline smoking status, and ICS use at baseline was generally consistent with the overall study population (Figure 3).
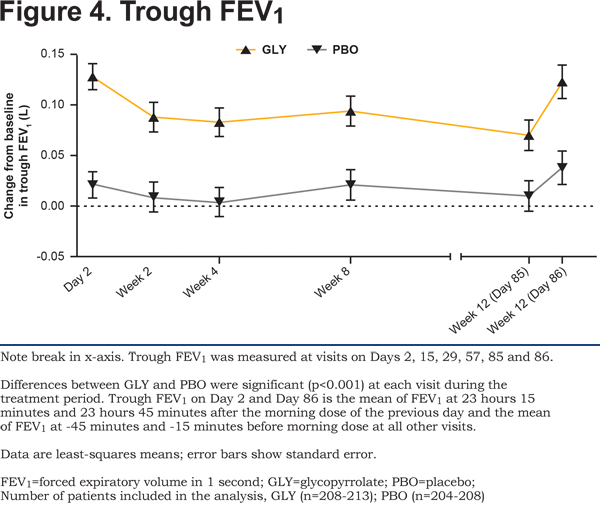
Superior bronchodilation with glycopyrrolate was supported by statistically significant improvement in trough FEV1 on Day 2 which was maintained at all timepoints through Week 12 (both Days 85 and 86) (Figure 4) confirming benefits with respect to trough FEV1 for twice-daily glycopyrrolate.
Glycopyrrolate showed an early onset of bronchodilation with statistically significant improvements in FEV1 at 5 and 15 minutes post-dose compared with placebo at Day 1 and Week 12 (all p<0.001; Figure 5A and 5B). Serial spirometry showed significantly higher improvements in FEV1 with glycopyrrolate versus placebo at all individual post-baseline timepoints from 0 to 12 hours during the study (p <0.001 at all timepoints; Figure 5A and 5B). Similarly, FVC at all timepoints from 0 to 12 hours post dose at Day 1 and Week 12 was significantly improved with glycopyrrolate versus placebo (data not shown).
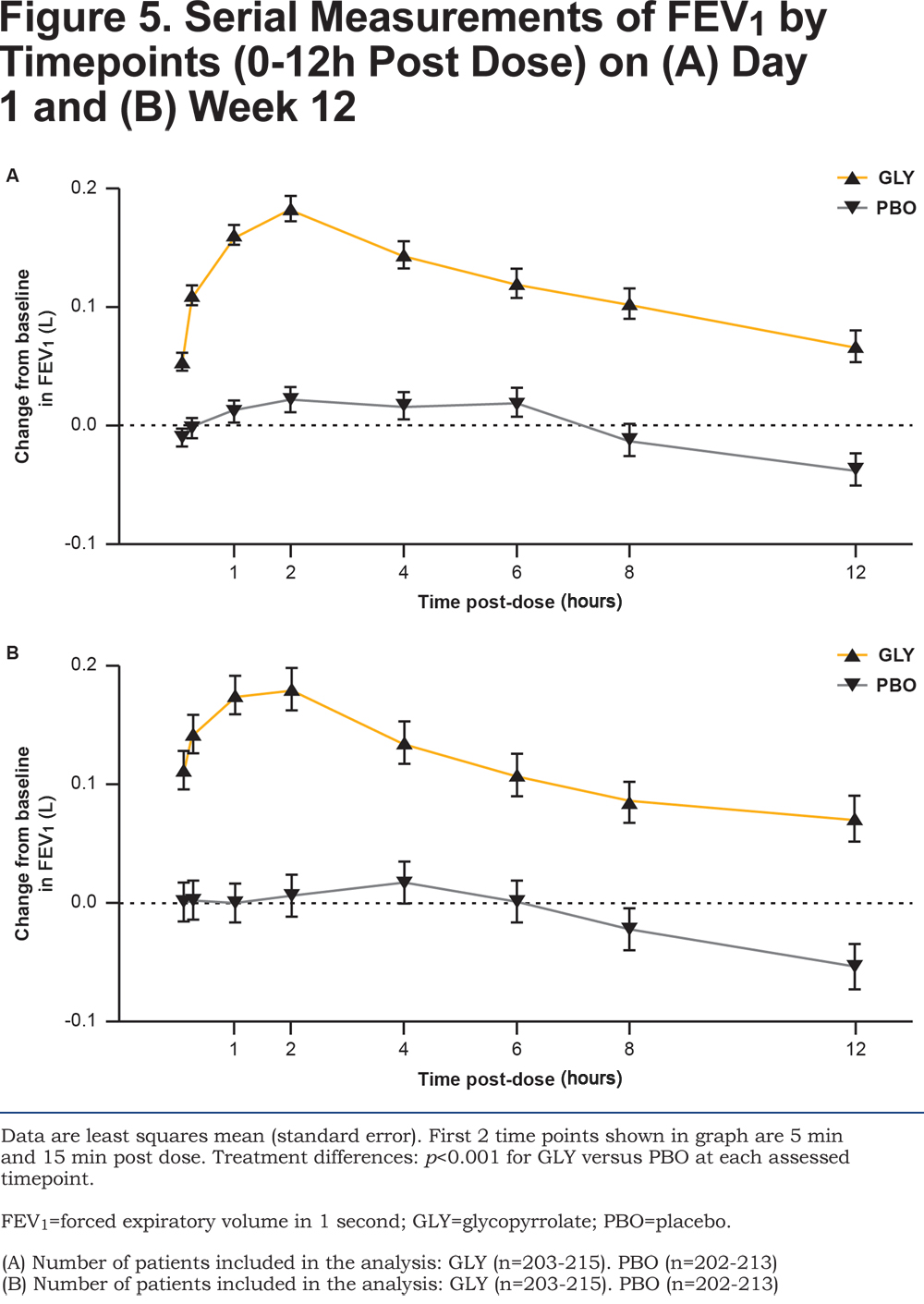
At Week 12, peak FEV1 and peak FVC for glycopyrrolate were significantly higher compared to placebo (peak FEV1 treatment difference 148 mL; p <0.001 and peak FVC treatment difference 201 mL; p <0.001; online supplementary material Table 1).
Health Status and Dyspnea
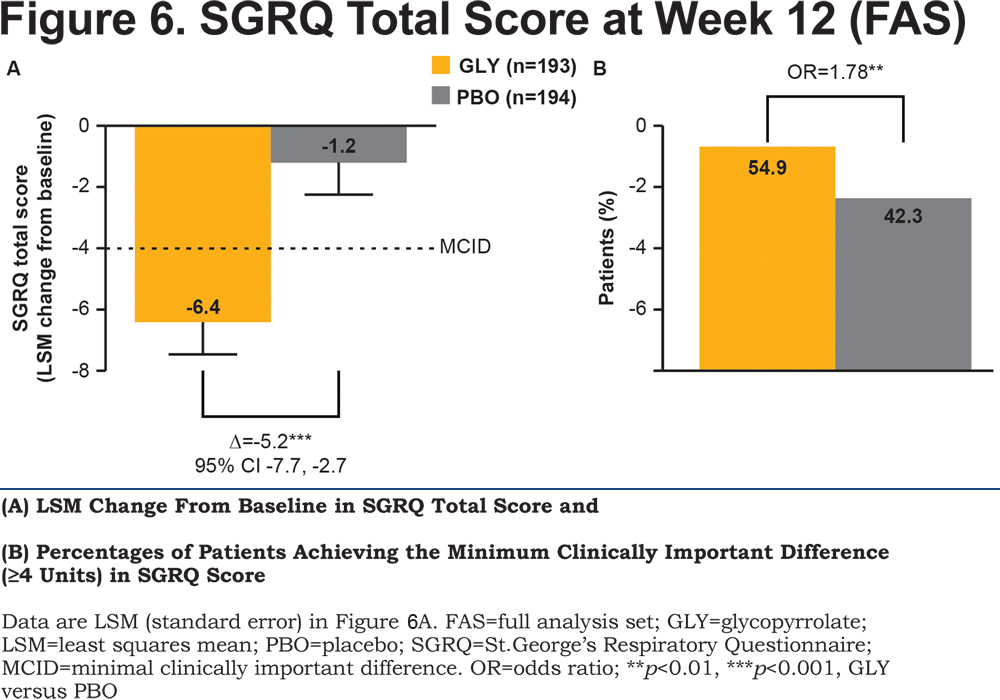
With respect to health status, glycopyrrolate showed a statistically significant and clinically relevant improvement in the SGRQ total score compared with placebo (p<0.001; Figure 6A) and 29.8% more patients in the glycopyrrolate group achieved a clinically relevant ≥4 units improvement versus placebo (p<0.01; Figure 6B).
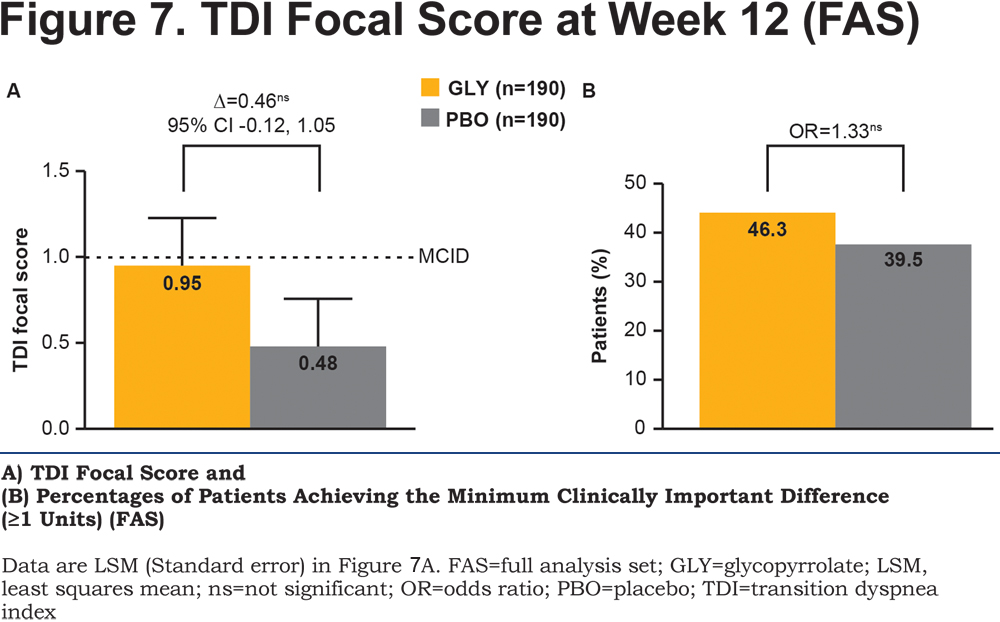
The TDI focal score showed a numerical reduction in dyspnea with glycopyrrolate compared with placebo (Figure 7A) but the difference was not statistically significant. Overall, more patients receiving glycopyrrolate (46.3%) achieved the MCID of ≥1 unit in dyspnea than those receiving placebo (39.5%; Figure 7B). The CAT score significantly improved by -1.5 units at Week 12 (online supplementary material Table 1).
Rescue Medication Use and Symptom Scores
The change from baseline in the use of rescue medication was significantly lower in daily (treatment difference -0.53 puffs/day; p<0.05), daytime (treatment difference -0.30 puffs/day; p<0.05), and nighttime (treatment difference -0.25 puffs/night; p<0.05) number of puffs in patients treated with glycopyrrolate compared with placebo over the 12 week treatment period (online supplementary material Table 2). Glycopyrrolate also showed a significant improvement in the daily total symptom score (derived from data recorded in the patient e-diary) compared with placebo (online supplementary material Table 2).
Safety
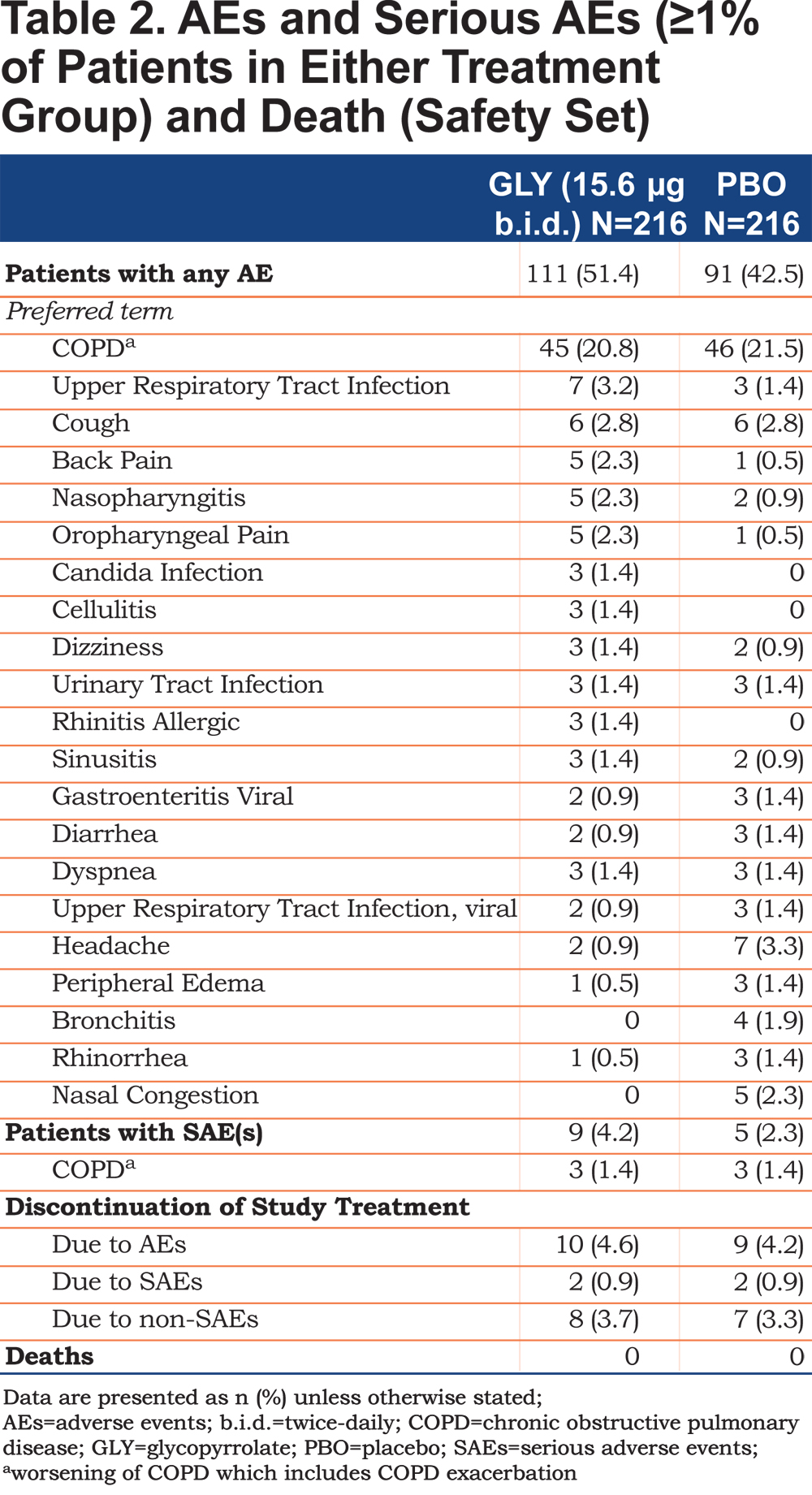
Adverse events were reported for 111 (51.4%) patients in the glycopyrrolate group versus 91 (42.5%) patients in the placebo group (Table 2). The incidence of serious AEs (SAEs) was 4.2% with glycopyrrolate versus 2.3% with placebo (Table 2). The majority of SAEs were respiratory-related, and the incidence rate was the same across both treatment groups (1.9% in both the treatment groups). COPD (exacerbation) was the most commonly reported AE, and was comparable across the treatment groups (glycopyrrolate 20.8% versus placebo 21.5%; Table 2). Oropharyngeal related AEs (upper respiratory infection, nasopharyngitis, and oropharyngeal pain) that are commonly seen with orally inhaled LAMAs, occurred at low frequencies in both groups (Table 2). Potential anti-cholinergic AEs occurred infrequently with less than 1.5% of patients reporting urinary tract infections (Table 2) and less than 1% reporting dry mouth, constipation and bladder outflow obstruction and urinary retention. The majority of AEs reported during the treatment period were of mild or moderate severity; 5.6% of patients in the glycopyrrolate group and 2.8% in the placebo group experienced severe AEs. No death occurred in either group. The proportion of patients who discontinued the study treatment due to AEs (with COPD being the most common AE [≥1% in any group] leading to discontinuation) and SAEs was similar between the treatment groups (Table 2). An adjudicated serious CCV AE was reported for 1 patient in the glycopyrrolate group only; this event was adjudicated as MACE and classified as a non-fatal myocardial infarction. No patient in the placebo group had an adjudicated serious CCV event. Atrial fibrillation/flutter events occurred in 4 patients (1.9%) in the glycopyrrolate group compared to 1 patient (0.5%) in placebo, of which new onset events were reported by 2 patients (0.9%) in the glycopyrrolate group versus none in the placebo group. The proportion of patients with newly occurring or worsening clinically notable Fridericia’s corrected QT interval (QTcF) values was similar between the treatment groups. One patient in the glycopyrrolate group had a QTcF value >480 msec. No meaningful differences between treatment groups were seen for laboratory evaluations and clinically notable vital signs.
Discussion
Bronchodilation with a LAMA and/or LABA is central to the management of COPD.1 As per the current clinical guidelines, LAMAs are recommended as a potential first choice therapy, alone or in combination, for patients at high risk of exacerbations.1 Glycopyrrolate 15.6 µg twice-daily provides an additional LAMA option which has proven to be clinically effective with an early onset of bronchodilatory action and is generally well tolerated with a low incidence of anticholinergic effects, as shown in this study. The overall results of the GEM2 study demonstrate that twice-daily glycopyrrolate is efficacious and safe compared with placebo in patients with moderate-to-severe COPD. The selection of the glycopyrrolate dose used in this study was based on the findings of a dose-ranging study in which glycopyrrolate 15.6 µg b.i.d. was demonstrated as an effective dose that showed clinically meaningful improvement in trough FEV1 versus placebo.8
The study met its primary objective with glycopyrrolate demonstrating superiority to placebo in terms of FEV1 AUC0-12h at Week 12. The magnitude of improvement (glycopyrrolate versus placebo) was 123 mL, which was considered clinically meaningful thus supporting the efficacy of glycopyrrolate 15.6 µg b.i.d. Glycopyrrolate was also statistically significantly superior to placebo for this endpoint on Day 1. Other secondary lung function parameters supported the primary endpoint with significant improvements in trough FEV1, pre-dose trough FEV1, peak FEV1, peak FVC, and FVC AUC0-12h. Bronchodilation with glycopyrrolate was evident at Day 1 and was maintained over the 12-week treatment duration. Compared to the GLOW14 and GLOW25 studies, generally, similar improvements in lung function were seen with glycopyrrolate versus placebo in this study at Week 12 (least square mean [LSM] treatment difference [trough FEV1] 108 to 97 mL in GLOW1 and GLOW2 studies [12-week data], respectively, versus 86 mL in this study). Notably, glycopyrrolate in this study has shown similar lung function improvement compared to other twice-daily LAMAs such as aclidinium in the ACCORD COPD II trial (change from baseline in trough FEV1 was 51 and 72 mL versus placebo at doses of 200 µg and 400 µg respectively).11 Of note, patient baseline characteristics, disease history and severity were comparable between the glycopyrrolate and placebo groups.
As per the GOLD guidelines, an improvement in symptoms is an important goal in the disease management of patients with COPD.1 Clinical guidelines and systematic reviews suggest that the effectiveness of COPD treatments should not be assessed by lung function alone but should also include a variety of other measures, in particular patient-reported outcomes such as dyspnea, health related quality of life and improved exercise endurance.1,12,13 Also, there is clear evidence that improvements in mean trough FEV1 is associated with improvements in SGRQ.14 In this study, glycopyrrolate showed significant improvements in health status, mean daily total symptom scores and mean daily rescue medication use over 12 weeks’ treatment and these were generally consistent with the known efficacy results of glycopyrrolate from other clinical trials.15 The reduced usage of rescue medication in patients receiving glycopyrrolate compared with placebo also suggests an improvement in symptoms. This overall improvement was also reflected in the SGRQ total scores which correlated well with symptom and rescue medication parameters. MCID for the SGRQ is a reduction of 4 units in the total score and is well established.16 In this study, the proportion of patients who achieved the MCID in the SGRQ total score was significantly higher with glycopyrrolate than with placebo. In addition, CAT was also used as an exploratory objective to evaluate the improvement in health status and it was observed that glycopyrrolate significantly improved the CAT score compared with placebo. With regards to dyspnea, there was a trend towards improvement in TDI focal score which might have achieved significance with longer study duration.
The safety profile of glycopyrrolate 15.6 µg b.i.d observed in this study was consistent with the known safety profile of glycopyrrolate.5,7,8,17,18 Anti-muscarinic side effects, such as dry mouth, constipation, urinary retention, and urinary tract infections, occurred with a low frequency in both the treatment groups. Overall, glycopyrrolate 15.6 μg b.i.d. was generally well tolerated over a treatment period of 12 weeks. In spite of some minor imbalances in the frequency of some individual AEs, no clinically relevant differences between the groups could be observed with respect to any type of AE. In patients with moderate-to-severe COPD, it had been previously demonstrated that glycopyrrolate was well tolerated and had a similar incidence of MACEs, when compared with placebo or active treatments.4,7 Similarly, in this study, based on the adjudicated findings, there was no imbalance in MACEs between the treatment groups and no deaths were observed in either group. The incidence of atrial fibrillation/flutter was low and comparable to that reported with other marketed LAMAs such as umeclidinium and tiotropium.19
The study has certain limitations. First, it should be considered that the study duration of 12 weeks with the current sample size was not powered for analysis of exacerbations or for the assessment of some of the patient-reported outcomes. Similarly, the long-term safety profile could not be established in a 12-week study. Furthermore, no active comparator arm was included. Longer duration studies or studies enriched for COPD exacerbations would be needed to further clarify effects on exacerbations and long term safety findings.
Conclusion
The results from the GEM2 study demonstrated that twice-daily glycopyrrolate 15.6 µg showed an early onset of action, apparent within 5 and 15 minutes post-dose and sustained 24-hour bronchodilation over 12 weeks compared with placebo. Glycopyrrolate also showed statistically significant improvements in COPD symptoms, health status, and rescue medication use. Overall, glycopyrrolate 15.6 µg b.i.d. was generally well tolerated in patients with moderate-to-severe COPD.
Acknowledgements
This study was sponsored by Novartis Pharma AG. The authors thank the patients who participated, and the staff at the participating clinical centers. The authors also thank Novartis clinical team members Fernando Mota, Pamalar Lewis, Cathy Surdouski, Colleen Spotts, Michael Kleinhoffer and Janet Jenkins for their contributions in executing the study and Vivek Khanna (professional medical writer; Novartis) for assistance in the preparation of this paper.
Declaration of Interest
EK has served on speaker panels, advisory boards and as consultant for AstraZeneca (Pearl), Amphastar, Forest Laboratories, Sunovion, Boehringer Ingelheim, Teva Labs, Novartis, Mylan and Theravance. TMS has received research funding from Novartis, GlaxoSmithKline, Boehringer Ingelheim, Forest Research Institute, Astra Zeneca, Sunovion, and Pearl Therapeutics. PK has received research support for studies of COPD from Astra Zeneca, Amphastar, Boehringer Ingelheim, Forrest Laboratory, Novartis, Mylan, Sunovian and Teva. AW has no conflict of interests. JHE, FP and PD are employees of the study sponsor, Novartis. MH is contracted to work for Novartis. All authors participated in the development and writing of the manuscript and take full responsibility for the content of the article. All authors approved the final draft that was submitted. PD, JHE, MH and FP contributed to the design of the study. EK, the principal investigator of the study, has read and commented on the full study report, and had final responsibility for the decision to submit for publication. TMS, PK and AW as investigators of the study, contributed to the writing of each draft of the manuscript. All the investigators were involved in primary data collection. PD, JHE, MH and FP, as employees of the sponsor, contributed to the design and preparation, conduct, analysis and interpretation of the study for the manuscript, and also contributed to the writing of each draft of the manuscript.