Running Head: Long-term Safety and Efficacy with IND/GLY in COPD
Funding Support: Funding for this project was provided by Novartis Pharmaceuticals, Corp.
Date of Acceptance: May 18, 2016
Abbreviations: indacaterol, IND; glycopyrrolate, GLY; chronic obstructive pulmonary disease, COPD; twice daily, B.I.D; once daily, O.D.; adverse event, AE; forced expiratory volume in 1 second, FEV1; major adverse cardiovascular events, MACE; cardiovascular, CV; inhaled corticosteroids, ICS; long-acting beta2-agonists, LABA; long-acting muscarinic antagonist, LAMA; fixed dose combination, FDC; Food and Drug Administration, FDA; Global initiative for chronic Obstructive Lung Disease, GOLD; forced vital capacity, FVC; modified Medical Research Council, mMRC; Interactive Response Technology, IRT; electrocardiogram, ECG; serious adverse event, SAE; cerebrovascular, CCV; Medical Dictionary for Regulatory Activities, MedDRA; mixed-model repeated-measures, MMRM; full analysis set, FAS; linear mixed model, LMM; COPD assessment test, CAT; hazard ratio, HR
Citation: Ferguson GT, Taylor AF, Thach C, et al. Long-term maintenance bronchodilation with indacaterol/glycopyrrolate versus indacterol in modertate-to-severe COPD patients: The Flight3 study. Chronic Obstr Pulm Dis. 2016; 3(4): 716-728. doi: http://doi.org/10.15326/jcopdf.3.4.2016.0131
Introduction
Chronic obstructive pulmonary disease (COPD) is a progressive debilitating respiratory condition characterized by respiratory symptoms with persistent and progressive airflow limitation.1 It is a major cause of poor health and death worldwide and contributes significantly to health care costs and comorbidity.2-4 COPD remains a significant public health challenge and is one of the leading causes of morbidity and mortality in the United States, affecting an estimated 27 million Americans.5 It is currently the third leading cause of death in the United States and claimed 134,676 lives in 2010.6 It results in 1.5 million visits to emergency departments, over 15 million physician visits every year and 739,000 hospitalizations.7,8
COPD is a treatable disease. However, due to the heterogeneity of the disease and its response to current therapies, new therapeutic options are needed. Current COPD treatment guidelines recommend the use of one or more bronchodilators, depending on the severity of patients’ airflow limitations, history of exacerbations, and symptoms.1 Inhaled bronchodilator therapies, such as selective beta2-adrenergic receptor agonists and muscarinic receptor antagonists, are widely used treatment options for COPD, as are inhaled corticosteroids (ICS) in combination with a long-acting beta2-agonists (LABA).1 Studies have demonstrated that complementary mechanisms of action of a LABA and a long-acting muscarinic antagonist (LAMA) significantly improve bronchodilation in COPD patients compared with the respective monotherapy components.9,10 Combining bronchodilators, each with a different mechanism of action to influence airflow limitation, may maximize the bronchodilator response and impact the inter- and intra-patient variability in bronchometer tone associated with COPD.11,12 In addition, several studies have demonstrated that LABA/LAMA combinations have a better impact on lung function, inspiratory capacity, and quality of life compared with the respective monotherapies.13-15 Indacaterol maleate/glycopyrrolate (IND/GLY; QVA149) is a fixed-dose combination (FDC) of a LABA (indacaterol [IND]; QAB149) and a LAMA (glycopyrrolate [GLY]; NVA237) intended to be used for the long-term maintenance treatment of airflow obstruction in patients with COPD including chronic bronchitis and/or emphysema. The safety and efficacy of both IND and GLY have been demonstrated, with each monocomponent having a safety profile similar to that of a placebo.16-18
FDC therapy may also offer a potential advantage over monotherapy by increasing treatment adherence through the use of a single inhaler. Patients using multiple inhalers have demonstrated higher risk of exacerbations along with significantly higher health care costs and utilization.20 IND/GLY provides the opportunity for patients with COPD to receive dual bronchodilator therapy using 1 device, which results in rapid bronchodilation and a sustained efficacy for 12 hours.21 In December 2013, the Food and Drug Administration (FDA) approved the first dual long-acting bronchodilator (Anoro® Ellipta®) for the long-term maintenance treatment of airflow obstruction in patients with COPD including chronic bronchitis and emphysema. Stiolto™ Respimat®, a once-daily (o.d.) FDC of tiotropium and olodaterol, was approved in June 2015 by the FDA for treatment of COPD. In addition, the FDA has recently approved the dual combination (IND/GLY) Utibron Neohaler™ for the long-term maintenance treatment of airflow obstruction in patients with COPD.
The purpose of the FLIGHT3 study was to demonstrate the long-term safety and tolerability of twice-daily (b.i.d.) IND/GLY compared with an active comparator, IND, administered o.d. in patients with moderate-to-severe COPD.
Methods
Study Design
FLIGHT3 was a multicenter, randomized, double-blind, parallel-group study that compared safety and tolerability over a 52-week period (ClinTrials.gov identifier: NCT01682863). The first patients were enrolled on October 26, 2012, and the study was completed on June 30, 2014. Patients were screened across 88 centers in 6 countries: Bulgaria (5), Finland (4), Hungary (10), Romania (10), Spain (8), and the United States (51). The study complied with International Conference on Harmonisation (ICH) Harmonised Tripartite Guidelines for Good Clinical Practice, applicable local regulations, and ethical principles laid down in the Declaration of Helsinki.
Patients
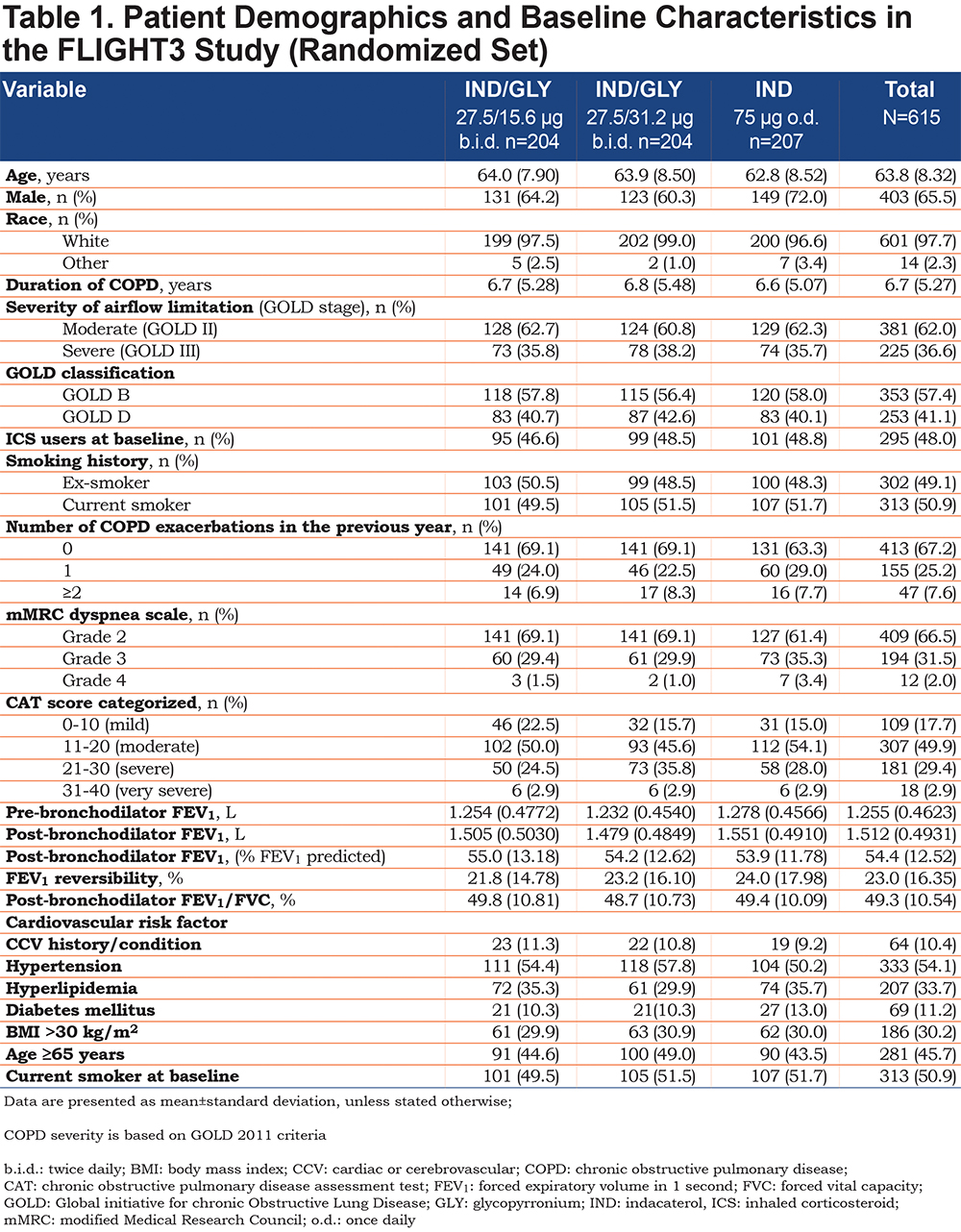
The study recruited male and female patients aged ≥40 years who had stable COPD according to the 2011 Global initiative for chronic Obstructive Lung Disease (GOLD) criteria.1 Patients were included if they had moderate-to-severe airflow limitation, as indicated by post-bronchodilator forced expiratory volume in 1 second (FEV1) ≥30% and ˂80% of the predicted normal and a post-bronchodilator FEV1/forced vital capacity (FVC) ratio ˂0.70 at run in. The patients were either current or ex-smokers, with a smoking history of at least 10 pack years, and were symptomatic, as defined by a modified Medical Research Council (mMRC) dyspnea scale, Grade ≥2.
Patients with any history of asthma or concomitant pulmonary disease or with a significant disease other than COPD that could significantly confound the trial results or preclude trial completion (including cardiovascular [CV], neurological, endocrine, immunological, psychiatric, gastrointestinal, hepatic, or hematological abnormalities) were excluded. Patients were also excluded if they had a COPD exacerbation that required treatment with antibiotics and/or systemic corticosteroids and/or hospitalization in the 6 weeks prior to Visit 1. Further information on exclusion criteria is available in the online supplementary data (eTable 1). All patients provided written informed consent, and all protocols were approved by an ethics committee.
Study Treatments and Randomization
This study comprised a 7-day screening period and a 14-day run-in period, followed by randomization 1:1:1 to receive IND/GLY (27.5/15.6 µg b.i.d.), IND/GLY (27.5/31.2 µg b.i.d.), or IND (75 µg o.d.). All treatments were delivered via the Neohaler™ device. The randomized, multicenter, double-blind design of this study was selected to provide rigor and reduce bias in addressing the long-term safety of the FDCs IND/GLY 27.5/15.6 and 27.5/31.2 μg b.i.d. versus IND 75 μg o.d. A placebo group was not included because of ethical concerns based on the severity of patients studied and the study duration. The patients were treated for 52 weeks in order to establish the long-term safety of IND/GLY. The inclusion of IND 75 μg o.d. as a comparator allowed for the comparison of the safety profile of IND/GLY with that of IND, an approved treatment for COPD in the United States.22 A higher dose of the FDC IND/GLY 27.5/31.2 μg b.i.d, with the LAMA component being twice that of the FDC IND/GLY 27.5/15.6 μg b.i.d., was included as a treatment group to obtain additional safety data. The dose and dosing regimen of IND are based on the approved dose in the United States and supported by a previous study (NCT01959412). The dose and dosing regimen of GLY are based on an initial dose-ranging study (NCT00501852),22 which investigated o.d. doses of GLY, and an additional dose-ranging study (NCT01119950),23 which included multiple o.d. and b.i.d. doses.
All eligible patients were randomized via Interactive Response Technology (IRT) to one of the treatment groups. Treatment randomization was stratified based on the smoking status, ICS use, and severity of airflow limitation. Patients were instructed to take 1 capsule each morning (between 07:00 and 11:00 h) and 1 in the evening (between 19:00 and 23:00 h). Each patient was provided with a salbutamol/albuterol inhaler, which was permitted for use as rescue medication throughout the study. Nebulized salbutamol/albuterol was not permitted. Patients used an electronic diary (e-diary) to capture the use of rescue medication.
Outcomes and Assessments
The primary objective was to evaluate the safety and tolerability of IND/GLY (27.5/15.6 or 27.5/31.2 µg) versus IND (75 µg) in terms of adverse event (AE)-reporting rates in patients with moderate-to-severe COPD over 52 weeks. Secondary objectives were to evaluate the bronchodilator effect of IND/GLY (27.5/15.6 or 27.5/31.2 µg) versus IND (75 µg) on the mean FEV1 at pre-dose 15 and 45 minutes (pre-dose trough FEV1) at Week 52 and on FEV1 and FVC at all post-baseline time points (including post-dose 1 h). Other secondary objectives were to evaluate the safety and tolerability of IND/GLY (27.5/15.6 or 27.5/31.2 µg) versus IND (75 µg) in terms of vital signs, electrocardiogram (ECG), laboratory evaluations, and treatment discontinuation and the effect of IND/GLY (27.5/15.6 or 27.5/31.2 µg) versus IND (75 µg) on time to first moderate or severe exacerbation, COPD symptoms reported, and the number of puffs/day of rescue medication during the 52-week treatment.
Sample Size and Statistical Analyses
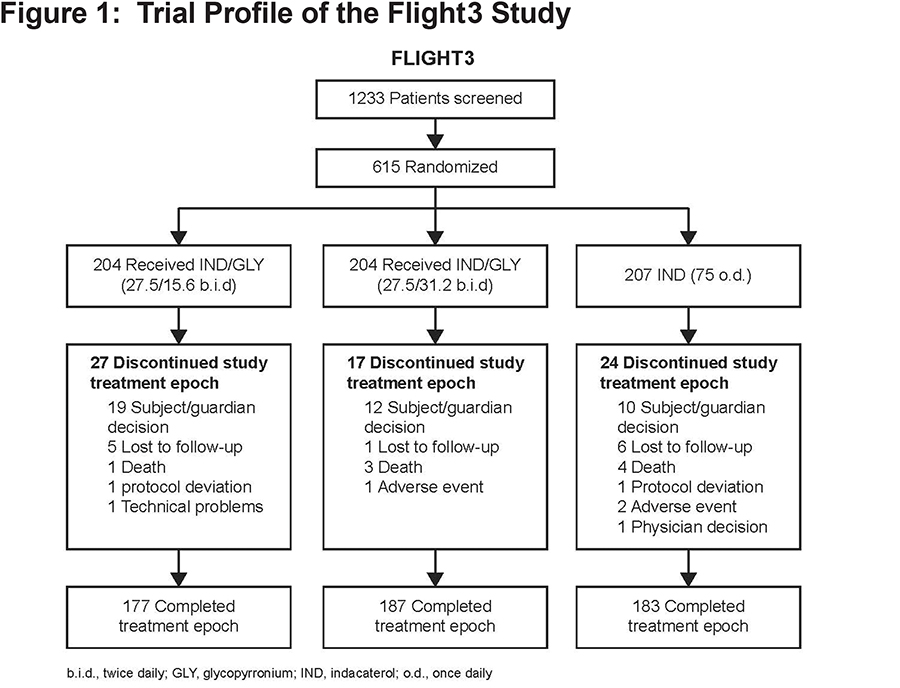
A total of 615 patients with a clinical diagnosis of COPD and moderate or severe airflow limitation were randomized (1:1:1) into the 3 treatment groups (Figure 1). The primary endpoint was analyzed based on the Safety set (all patients that received at least 1 dose of study drug and had at least 1 post-baseline safety assessment). The safety assessment included all safety measurements, such as AEs and COPD exacerbations. All treatment-emergent AEs starting on or after the time of the first administration of the study drug but not later than 7 days (30 days in the case of a serious adverse event [SAE]) after the last administration, were summarized. Serious cardiac or cerebrovascular (CCV) AEs and deaths were adjudicated by an independent adjudication committee. All AEs were coded according to the Medical Dictionary for Regulatory Activities (MedDRA; version 17.0). No statistical hypothesis testing was performed on AEs.
A composite endpoint of major adverse cardiovascular events (MACE) was included in the analysis, which represented all serious CCV AEs adjudicated by an independent committee to one of the following categories: a) non-fatal myocardial infarction, b) hospitalization for unstable angina, c) non-fatal stroke, d) heart failure requiring hospitalization, and e) coronary re-vascularization (coronary artery bypass graft or percutaneous coronary intervention).
Particular attention was paid to the LABA and LAMA drug classes in terms of safety variables, such as increased heart rate, increased blood pressure, CCV (major cerebrovascular events, cardiac arrhythmias), hypokalemia, diabetes and hyperglycemia, corrected QT interval prolongation, paradoxical bronchospasm, narrow-angle glaucoma, blurred vision, urinary outflow obstruction, urinary retention, and anticholinergic syndrome.
The change from baseline in pre-dose trough FEV1 from Week 4 to Week 52 was analyzed using a mixed-model repeated-measures (MMRM) analysis on a full analysis set (FAS; all randomized patients who received at least 1 dose of study medication). This model included treatment, baseline FEV1, visit, treatment by visit interaction, visit by baseline FEV1 interaction, smoking status at baseline, baseline ICS use, airflow limitation severity, and region as fixed effects with unstructured variance-covariance error matrix. Similar analysis was performed for change from baseline in post-dose 1-h FEV1 (as measured at the same visits and at Day 1), pre-dose trough FVC, and post-dose 1-h FVC.
The time to first COPD exacerbation was assessed using Kaplan-Meier curve and analyzed using Cox regression model for the FAS. The rate of COPD exacerbations was analyzed using a generalized linear model assuming a negative binomial distribution. The log (exposure time) was used as an offset variable in the model.
Daily, morning, and evening symptom scores were analyzed using a linear mixed model (LMM). A similar LMM was also used to analyze daily, daytime, and nighttime rescue mediation uses. The change from baseline in COPD assessment test (CAT) total score at Weeks 12, 28, 44, and 52 was analyzed using a similar MMRM as that used for pre-dose trough FEV1, with baseline FEV1 replaced by baseline CAT total score.
Results
Patients
Of the 615 patients randomized to treat, 85.2% completed the planned study treatment. One randomized patient in the IND group did not receive the study medication and was excluded from the analysis. Overall, 68 patients discontinued the treatment, with the most frequent reason for discontinuation being patient/guardian decision (n=41) (Figure 1). Other reasons for treatment discontinuation included lack of efficacy, protocol deviations, and physician’s decision (with comparable rates among treatment groups) (Figure 1). Most patients (>99%) were compliant with the study medication per protocol (which categorized compliance as 80%-100%) with a similar rate between the twice daily and once daily treatment groups.
Patient demographics were comparable between the 3 treatment groups (Table 1).The majority of the patients were male, and nearly all patients were white. (Approximately 60% of patients were classified as Group B and approximately 40% as Group D according to GOLD 2011 strategy24 (Table 1). Since an mMRC score of ≥2 was required for study entry, no Group A or C patients were included. In the year prior to enrollment, only 33% patients had a history of COPD exacerbation (Table 1).
Outcomes
Adverse Events
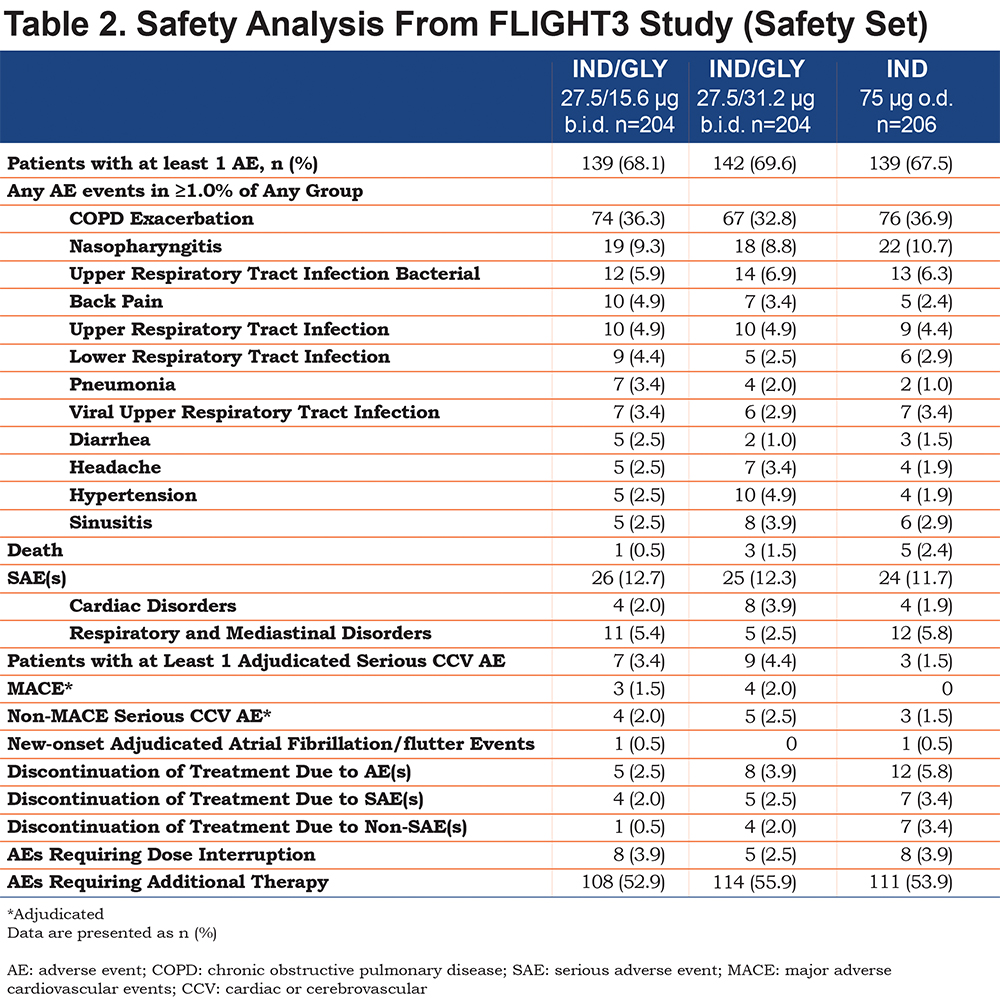
The incidence of AEs was similar across the treatment groups (IND/GLY 27.5/15.6 µg, 68.1%; IND/GLY 27.5/31.2 µg, 69.6%; IND 75 µg, 67.5%) (Table 2). Most of the reported AEs were mild (22.1%-25.7%) or moderate (30.6%-35.3%) in severity. COPD exacerbation was the most frequently occurring AE, and the AE rate was similar across the treatment groups (IND/GLY 27.5/15.6 µg, 36.3%; IND/GLY 27.5/31.2 µg, 32.8%; IND 75 µg, 36.9%) (Table 2).
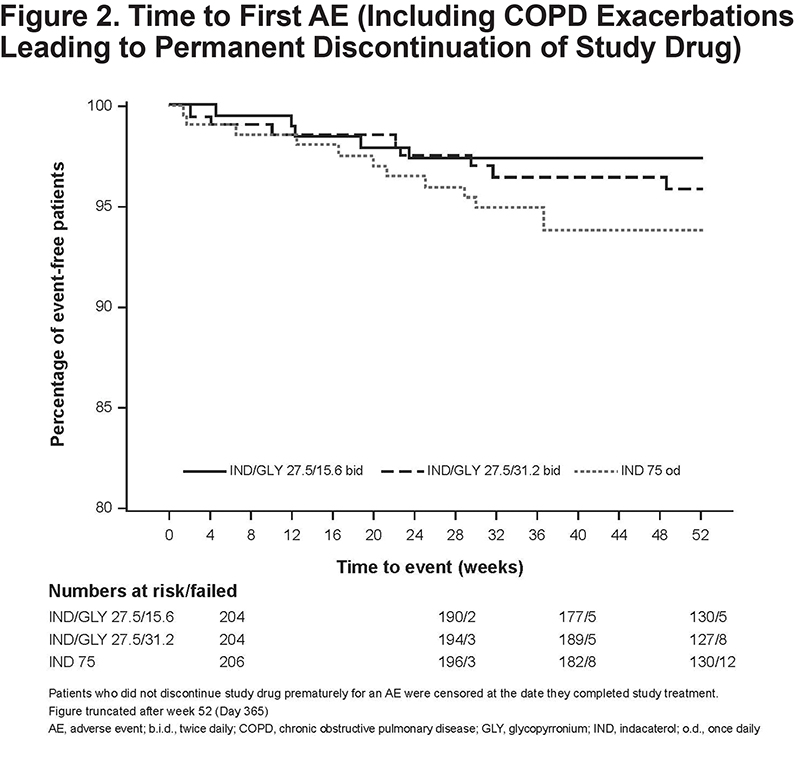
The rate of discontinuation from the study treatment due to AEs was lowest for IND/GLY 27.5/15.6 µg (2.5%), followed by IND/GLY 27.5/31.2 µg (3.9%) and IND 75 µg (5.8%). The 3 treatment groups demonstrated comparable trends when assessing the time to first AEs leading to permanent discontinuation (Figure 2). The incidence of drug-related AEs was higher with IND 75 µg (9.2%) than that with IND/GLY 27.5/31.2 µg (6.9%) or IND/GLY 27.5/15.6 µg (4.4%); no specific pattern of drug-related discontinuations was observed.
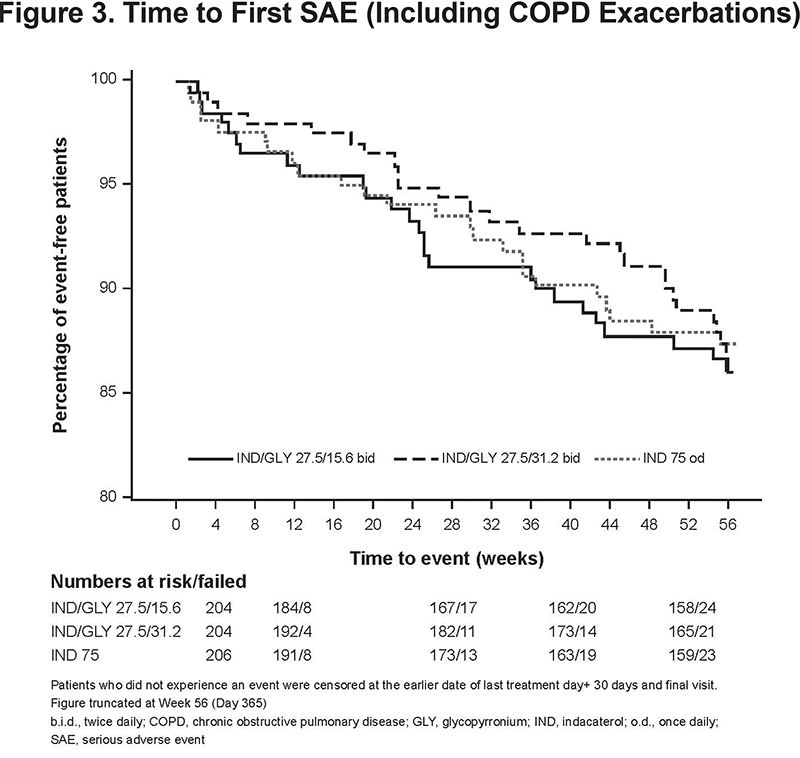
The incidence of SAEs was low and comparable between all treatment groups: IND/GLY 27.5/15.6 μg, 12.7%; IND/GLY 27.5/31.2 μg, 12.3%; and IND 75 μg, 11.7% (Table 2). Similar trends were observed between the treatment groups when assessing the time to first SAE (including COPD exacerbation) (Figure 3). The rate of discontinuation of the study medication due to SAEs was also low and comparable between all treatment groups (IND/GLY 27.5/15.6 µg, 2.0%; IND/GLY 27.5/31.2 µg, 2.5%; IND 75 µg, 3.4%).
The incidence of CCV events as a risk category was comparable between IND/GLY 27.5/15.6 µg (4.4%) and IND 75 µg (4.4%) but was higher with IND/GLY 27.5/31.2 µg (7.4%). The incidence of new-onset atrial fibrillation/flutter events was comparable between IND/GLY 27.5/15.6 µg (0.5%) and IND 75 µg (0.5%), and a 0.0% event rate was reported for IND/GLY 27.5/31.2 ug. The incidence of any AEs of special interest (CCV and non-CCV) was comparable across the treatment groups (IND/GLY 27.5/15.6 μg, 50.5%; IND/GLY 27.5/31.2 μg, 52.9%; IND 75 μg, 47.6%). The most frequently reported AEs of special interest were COPD exacerbation (IND/GLY 27.5/15.6 μg, 36.3%; IND/GLY 27.5/31.2 μg, 32.8%; IND 75 μg, 36.9%) and AEs related to the respiratory composite endpoint of upper and lower respiratory tract infections, pneumonia, bronchitis (IND/GLY 27.5/15.6 μg, 21.1%; IND/GLY 27.5/31.2 μg, 23.0%; IND 75 μg, 23.8%).
The number of MACE and/or CV deaths were also comparable across the treatment groups: IND/GLY 27.5/15.6 µg, 4; IND/GLY 27.5/31.2 µg, 5; IND 75 µg, 3. Overall, 9 patients died during the study: IND/GLY 27.5/15.6 µg, 1; IND/GLY 27.5/31.2 µg, 3; IND 75 µg, 5. The majority of the deaths were cardiovascular-related. Importantly, only 1 death was reported over 52 weeks on the approved Utibron™ dose (IND/GLY 27.5/15.6 µg). None of the deaths were considered to be related to the study drug by the reporting investigator.
No clinically meaningful between-group differences for any clinical chemistry or hematological parameter were observed. ECG parameters did not demonstrate any meaningful between-group differences in the rates of abnormalities seen for Fridericia-corrected QT interval values and other ECG intervals. There was no evidence of a treatment-related effect on vital signs (pulse and systolic and diastolic blood pressures) (see online supplemental data eTables 2 and 3).
Trough FEV1 and FVC
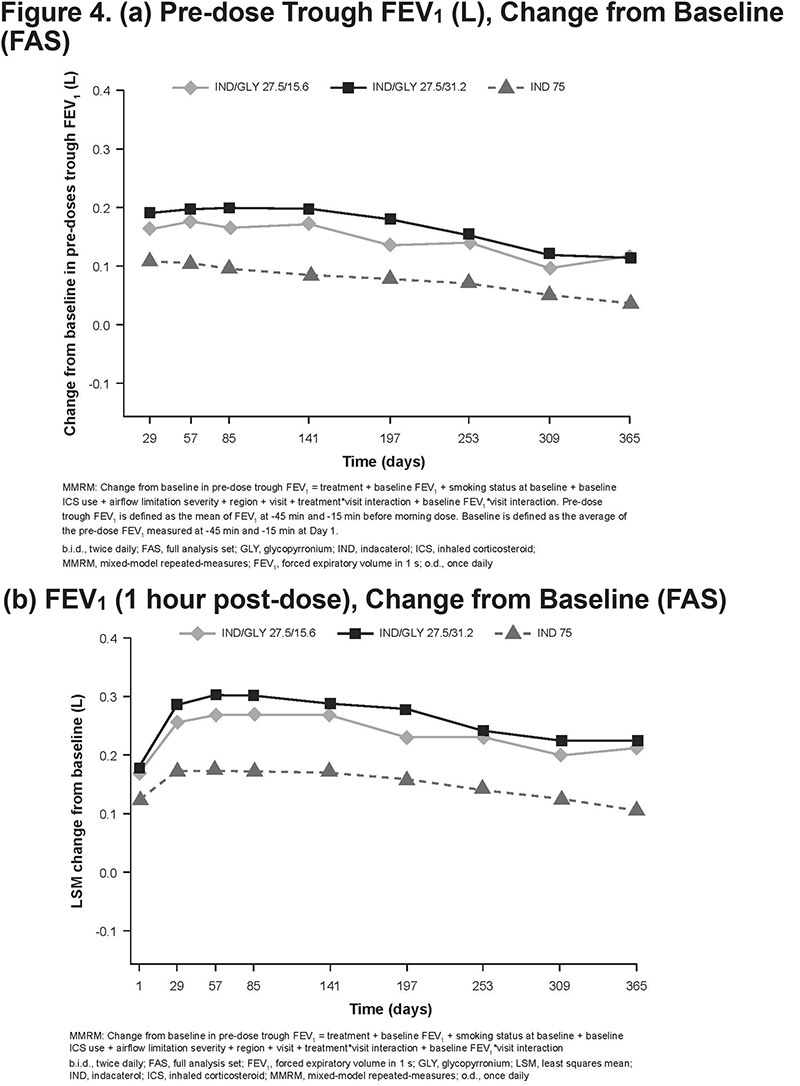
Improvements in pre-dose trough FEV1 with both the IND/GLY doses were consistently superior to those with IND at Week 52 (treatment difference, 80 and 79 mL for IND/GLY 27.5/15.6 and 27.5/31.2 µg, respectively) at all visits up to Week 52 (Figure 4 [a]). Improvements in post-dose 1-h FEV1 with IND/GLY 27.5/15.6 µg were consistently superior to those with IND 75 µg at all visits up to 52 weeks (∆, 44 mL on Day 1; ∆, 108 mL on Day 365) (Figure 4 [b]). Similarly, pre-dose trough FVC and post-dose 1-h FVC with both the IND/GLY doses were superior to those with IND over 52 weeks (online supplemental data eFigures 1 and 2).
COPD Exacerbation and Patient-reported Endpoints
The number of patients who experienced mild, moderate, or severe COPD exacerbations was comparable among the treatment groups. Both IND/GLY doses resulted in a 12% reduction in the risk of moderate or severe COPD exacerbation compared with IND, although this was not statistically significant (hazard ratio [HR], 0.88 and p=0.516 for IND/GLY 27.5/15.6 µg versus IND 75 µg; HR, 0.88 and p=0.502 for IND/GLY 27.5/31.2 µg versus IND 75 µg). There were numerical between-group differences in the rate of moderate or severe COPD exacerbations, with IND/GLY 27.5/15.6 µg demonstrating 25% fewer exacerbations versus IND 75 µg (rates ratio, 0.75 and p=0.163 for IND/GLY 27.5/15.6 µg versus IND 75 µg; rates ratio, 0.87 and p=0.464 for IND/GLY 27.5/31.2 µg versus IND 75 µg). A majority of symptom score endpoints, use of rescue medication, and CAT scores were similar between the treatment groups (online supplemental data eTables 4 and 5 and eFigure 3).
Discussion
In this 1-year study, the safety and efficacy of IND/GLY, a b.i.d. LABA/LAMA combination therapy, was compared with that of IND, a LABA, in COPD patients with moderate-to-severe airflow limitation. We believe that this is the first time that a 52-week LABA/LAMA (b.i.d) safety study is being reported in the United States. Overall, 14.8% of patients discontinued the study treatment. The range of discontinuation rates was lower than that observed in other 52-week COPD studies (30%-40%).25-26 The rate of discontinuation of the study treatments due to AEs was lowest for IND/GLY 27.5/15.6 µg b.i.d., followed by IND/GLY 27.5/31.2 µg b.i.d. and IND 75 µg o.d. Fewer patients on IND/GLY 27.5/15.6 µg b.i.d. discontinued treatment compared with those on IND 75 µg o.d.; however, this difference was not clinically significant. The incidence of MACE and/or CV deaths was also comparable across the treatment groups. The incidence of CCV events as a risk category was comparable between IND/GLY 27.5/15.6 µg b.i.d. and IND 75 µg o.d. and was higher with IND/GLY 27.5/31.2 µg b.i.d. The overall incidence of AEs and SAEs was similar between the treatment groups. There were no clinically meaningful differences in laboratory tests, ECG, and vital signs. A majority of symptom score endpoints, use of rescue medication, and CAT scores were similar between the treatment groups. The AEs, SAEs, and the overall safety profile of IND/GLY over 1 year were similar to those in other IND/GLY studies of 12-week durations.27
The safety profiles of FDCs of LABA/LAMAs are of key importance, particularly because earlier analyses examining the CV safety profile of these drug classes have raised questions related to their safety.28,29 This study contained 2 different doses of GLY to alleviate safety concerns typically associated with LAMA use.28,29 Both the doses of GLY (15.6 and 31.2 µg b.i.d.) were safe and well tolerated over 52 weeks, with no significant differences between the doses. A pooled safety analysis of a higher IND/GLY dose (110/50 µg o.d.) has also demonstrated that IND/GLY has a safety profile comparable to that of a placebo.30 Importantly, a shorter-term cardiovascular safety study also demonstrated that IND/GLY had a safety profile similar to that of placebo in terms of CV assessments 31 and that IND/GLY was well tolerated, with overall AE rates similar to those with a placebo.31 A pooled analysis of the 6-month safety data extrapolated from 4 trials (SHINE, ILLUMINATE, ENLIGHTEN, and ARISE) on the o.d. 110/50 µg dosage also demonstrated that the proportion of COPD patients treated with IND/GLY who experienced CCV events was similar to the proportion of such patients treated with tiotropium and lower than the proportion of such patients receiving placebo.32 Recently, the FLIGHT1 and FLIGHT2 studies have demonstrated that the safety and tolerability profile of IND/GLY in patients receiving IND/GLY 27.5/15.6 µg is comparable to those of the monocomponents IND and GLY or placebo after 12 weeks.27 Compared with the short-term FLIGHT1 and FLIGHT2 studies, the FLIGHT3 study demonstrated the long-term safety of IND/GLY versus IND over a longer treatment period (52-week).
The effect of IND/GLY on pulmonary function was evaluated as a secondary objective. In the pooled FLIGHT studies, statistically superior bronchodilation was observed with IND/GLY than that with its monocomponents or placebo.27 In this study, the 1-year efficacy data on pulmonary function again demonstrated that the patients receiving IND/GLY had a superior long-term bronchodilation compared with patients receiving IND. Pre-dose trough FEV1 and post-dose 1-h FEV1 were consistently superior with IND/GLY 27.5/15.6 μg versus IND 75 μg at all visits over 52 weeks, indicating maintenance of lung function and persistence of efficacy. Both the doses of IND/GLY achieved approximately 80mL in trough FEV1 versus IND alone. In addition, the onset of efficacy was quick as FEV1 (post-dose 1-h) was 44 mL on Day 1 and 108mL on Day 365 for IND/GLY versus IND alone.
Although this study was neither designed nor powered as an exacerbation trial with high-risk patients, there were numerical between-group differences in the rate of reduction of moderate or severe COPD exacerbations, with IND/GLY 27.5/15.6 µg demonstrating 25% fewer exacerbations as compared with IND 75 µg. In addition, both the IND/GLY doses resulted in a 12% reduction in the risk of moderate or severe COPD exacerbations as compared with IND. Although these exacerbation results were not statistically significant, they are suggestive of a possible added therapeutic benefit if an appropriately designed and powered study is to be performed.
There are limitations of this study that have to be acknowledged. This study had no placebo group due to the long-term nature of the study. It was considered unethical to recruit patients with moderate-to-severe COPD in a placebo group for a 52-week duration. No significant difference was observed between IND/GLY 27.5/15.6 µg and IND 75 µg in terms of exacerbations/symptoms possibly because both the treatments were highly active, and the study was not powered to demonstrate this.
In conclusion, the FLIGHT3 study demonstrated the long-term safety, tolerability, and sustained efficacy of IND/GLY 27.5/15.6 µg b.i.d. in patients with moderate-to-severe COPD over 52 weeks of treatment. This data demonstrates that IND/GLY b.i.d. is a safe and well-tolerated treatment option for long-term use in patients with COPD.
Acknowledgements
The authors thank Sinead Flannery and David Bergin for their assistance in the preparation of the manuscript. Authors’ contributions:GF, AFT, AS-T, FP and DB participated in study design and concept of the study. Statistical analyses were overseen by CT and QW. All the authors contributed to the acquisition and interpretation of data and also in drafting and revision of the manuscript.
Declaration of Interest
GF is supported in part by multiple pharmaceutical clinical research projects, including grants from Novartis Pharmaceuticals, Corp.AFT, AST, FP and DB are employees of Novartis Pharmaceuticals Corp.