Running Head: Formoterol Fumarate Novel MDI Dose Range
Funding Support: This study was funded by Pearl Therapeutics Inc., a member of the AstraZeneca Group.
Date of Acceptance: September 6, 2016
Abbreviations: formoterol fumarate, FF; metered dose inhaler, MDI; chronic obstructive pulmonary disease, COPD; forced expiratory volume in 1 second, FEV1; area under the curve between 0 and 12 hours, AUC0-12; hydrofluoroalkane, HFA; long-acting beta2-agonist, LABA; dry powder inhaler, DPI; pharmacokinetic, PK; forced vital capacity, FVC; electrocardiogram, ECG; American Thoracic Society, ATS; inhaled corticosteroid, ICS; beats per minute, bpm; liquid chromatography/tandem mass spectrometry, LC/MS/MS; maximum observed plasma concentration, Cmax; modified intent-to-treat, mITT; least squares mean, LSM; standard error of the mean, SEM; confidence interval, CI; analysis of variance, ANOVA; standard deviation, SD; long-acting muscarinic antagonist, LAMA;lower limit of quantification, LLOQ
Citation: Sethi S, Fogarty C, Hanania NA, et al. Efficacy of formoterol fumarate delivered by metered dose inhaler using Co-suspension™ delivery technology versus Foradil® aerolizer® in moderate-to-severe COPD: a randomized, dose-ranging study. Chronic Obstr Pulm Dis. 2017; 4(1): 21-33. doi: http://doi.org/10.15326/jcopdf.4.1.2016.0158
Introduction
Improvements to existing drug delivery strategies, specifically inhaler technology, have the potential to substantially impact disease control in chronic obstructive pulmonary disease (COPD).1 Co-Suspension™ delivery technology is a novel formulation technique for enhancing the inhaled delivery of therapeutic agents.2 In this technology, the drug particles are suspended in hydrofluoroalkane (HFA) propellant by the use of spray-dried porous particles of distearoyl-phosphatidylcholine. This stable suspension is capable of delivering and aerosolizing very low doses of an active ingredient consistently, whether as individual agent or in combinations.3,4
Formoterol fumarate (FF) is a fast-acting and well-tolerated long-acting beta2-agonist (LABA), which has demonstrated improvements in lung function and quality of life versus placebo in patients with COPD.5-7 FF formulated using Co-Suspension delivery technology and delivered by pressurized metered dose inhaler (FF MDI) is under clinical development. In the first clinical study of FF MDI, exploring a range of doses (2.4, 4.8 and 9.6µg), the high dose (9.6µg) was superior to placebo and comparable to open-label Foradil® Aerolizer® 12μg8 (formoterol fumarate dry powder inhaler [DPI], hereafter Foradil®) in terms of both therapeutic efficacy and pharmacokinetic (PK) properties.9
The second study, which compared chronic administration (7 days) of FF MDI (7.2 and 9.6µg) with placebo and open-label Foradil® 12µg found that the 9.6µg dose was superior to placebo and non-inferior to Foradil® 12µg in terms of both therapeutic efficacy and PK.10 This current single-dose, cross-over, placebo-controlled study extends this initial dose-ranging investigation to compare the efficacy, safety and systemic exposure of 3 separate doses of FF MDI (7.2, 9.6 and 19.2μg) with that of Foradil® (12 and 24μg) in patients with moderate-to-severe COPD. The higher doses (FF MDI 19.2µg and Foradil® 24µg) were included to demonstrate separation of the higher dose from the lower dose(s) within each formulation.
Methods
Study Design
This was a randomized, double-blind (for FF MDI and placebo), placebo-controlled, cross-over, multicenter study, with an open-label active comparator, conducted at 3 sites in the United States between May and July 2011 (NCT01349868). Patients were randomized to 1 of 6 treatment sequences, each sequence included 6 study treatments: double-blinded FF MDI 7.2, 9.6, and 19.2µg (equivalent to 7.5, 10, and 20µg formoterol fumarate dihydrate, respectively); placebo MDI; and open-label Foradil® 12 and 24µg. Over a maximum period of 12 weeks, each patient received a single dose of each study treatment, separated by at least 3 days, in an order specified by a randomly generated sequence (Figure 1).
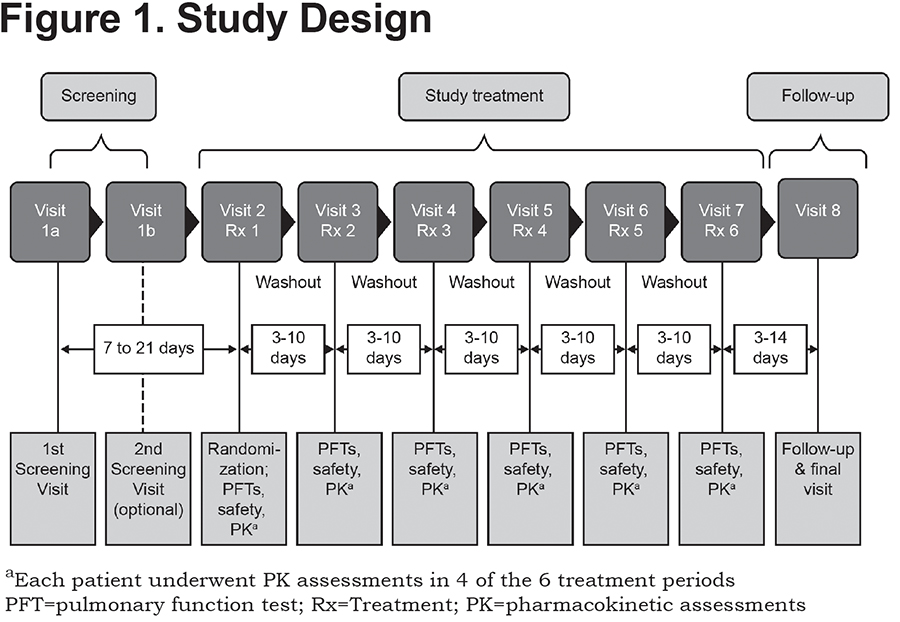
The study was conducted according to guidelines for Good Clinical Practice, International Conference on Harmonisation guidelines, the U.S. Code of Federal Regulations and the Declaration of Helsinki, and written informed consent was obtained from all patients prior to any screening assessment.11
Study Patient Population
Male and female participants, between 40 and 80 years of age at screening, with a diagnosis of COPD, airflow limitation (forced expiratory volume in 1 second/forced vital capacity [FEV1/FVC] ratio <0.7) and a smoking history of at least 10 pack years, were eligible for participation in the study. Patients were also required to have a post-bronchodilator FEV1 ≥30% and <80% of the predicted value and ≥750mL at screening (Visit 1) and pre-bronchodilator FEV1 <80% of the predicted value at baseline (Visit 2). In addition, patients had to demonstrate reversibility to a short-acting beta2-agonist (albuterol), defined as >12% and >150mL improvement in baseline FEV1 approximately 30 minutes following administration of 4 actuations of albuterol (Ventolin®) HFA or >200mL improvement in baseline FEV1 30 minutes following administration of 2 actuations of albuterol (Ventolin®) HFA. Participants were also required to have acceptable baseline laboratory tests and electrocardiogram (ECG) performed at screening. Chest X-rays or computed tomography scans dated within 6 months prior to screening were also examined for abnormalities.
Exclusion criteria included a primary diagnosis of asthma, alpha-1-antitrypsin deficiency or any other significant (including non-respiratory) condition. Patients who had lower respiratory tract infections that required antibiotics within 6 weeks prior to screening were excluded. Unstable or poorly-controlled COPD resulting in hospitalization during the 3 months prior to screening or requiring antibiotics or corticosteroids within 6 weeks before screening or between screening and baseline visits, also precluded participation in the study. Candidates with spirometry results that did not meet American Thoracic Society (ATS) standards or who required long-term oxygen therapy for >12 hours a day, were also excluded.
Participants taking certain prohibited COPD medications at screening (e.g., oral beta2-agonists, LABAs, corticosteroid/LABA combination products, phosphodiesterase inhibitors, cromoglycate/nedocromil inhalers, leukotriene antagonists or tiotropium), but who otherwise met study inclusion criteria, underwent washout periods of 7–21 days between screening and baseline visits. Patients receiving a maintenance dose of an inhaled corticosteroid (ICS) as part of a fixed-dose combination together with a LABA were switched to the corresponding ICS administered as a single agent, with short-acting bronchodilators (albuterol, ipratropium or albuterol/ipratropium), provided they have been maintained on a stable dose for at least 4 weeks. Protocol-adjusted ICS therapy was continued and remained stable for the duration of the trial. Patients receiving stable maintenance doses (>4 weeks) of an ICS monotherapy were permitted to continue.
Criteria for Discontinuation
A participant was discontinued from the study if any of the following changes were noted on 2 consecutive assessments conducted approximately 15 minutes apart, or at the discretion of the investigator: QT interval corrected for heart rate using Fridericia’s formula (QTcF) prolongation increase >60 milliseconds from test day baseline and >500 milliseconds at any time after taking study drug; heart rate increase >40 beats per minute (bpm) from test day baseline and >120bpm at any time after taking study drug; systolic blood pressure increase >40mmHg from test day baseline and >180mmHg at any time after taking study drug; FEV1 decrease >20% from test day baseline on 2 consecutive spirometry assessments obtained at least 15 minutes apart with associated symptoms of dyspnea.
Study Treatments
Three doses of FF MDI were assessed (7.2, 9.6 and 19.2μg); each administered as 2 actuations of the FF MDI containing half of the target dose per actuation. Two doses of Foradil® were administered as the active comparator, 12 and 24µg, equivalent to 1 and 2 capsules, respectively, inhaled via the DPI. Investigators and participants were blinded to FF MDI and placebo MDI treatment using non-distinguishable MDIs. Blinding with regard to Foradil® was not performed. Albuterol sulfate (Ventolin®) HFA could be administered as a rescue medication during treatment visits on an as-needed basis and according to the discretion of the Investigator.
Study Assessments
Participants were randomized to treatment sequences at baseline (Visit 2). Subsequent clinic visits were spaced between 3 and 10 days after the administration of each study treatment in the sequence, and 3–14 days between the last study treatment and the final follow-up visit (Visit 8). Spirometry was performed at all treatment visits at 60 and 30 minutes prior to the administration of study treatment. The average of these 2 readings was used to establish test-day baseline measurements for FEV1 and FVC. Following study drug administration, spirometry was obtained at 0.25, 0.5, 1, 2, 4, 6, 8, 10, 11.5 and 12 hours post-dosing. All sites used identical spirometry systems (KoKo Spirometer, nSpire Health, Inc., Louisville, Colorado) and customized, study-specific software.
Safety assessments, including physical examination, vital sign measurement, 12-lead ECG and clinical laboratory assessments, were performed at all study visits, including the follow-up visit 3–14 days after the final treatment. ECG measurements and blood samples for laboratory analyses were taken before and after the administration of study treatment. Adverse events were also recorded, with tremor and paradoxical bronchospasm summarized separately. Paradoxical bronchospasm was defined as a reduction in FEV1 of >20% from test-day baseline with associated symptoms of wheezing, shortness of breath, or cough.
Systemic Exposure
Blood samples for PK analysis were obtained from each patient 30 minutes prior to study drug administration, at 2, 6 and 20 minutes and at 1, 2, 4, 8, 10 and 12 hours post-dose. Samples were taken on 4 treatment days for each participant; during FF MDI 9.6µg and Foradil® 12µg treatment periods, and at 2 randomly selected periods, as determined by the Interactive Web Response System. Plasma levels of FF were determined using a validated high performance liquid chromatography/tandem mass spectrometry (LC/MS/MS) method.
Study Endpoints
The primary efficacy endpoint was the FEV1 area under the curve between 0 and 12 hours (FEV1 AUC0-12) normalized by dividing by the time relative to dosing of the last assessment (12 hours). Secondary efficacy endpoints included the peak change from baseline in FEV1 and change from baseline at 12-hour post-dose trough FEV1, defined as the mean of FEV1 assessments taken at 11.5 and 12 hours post-dose. Efficacy measurements were defined according to ATS/European Respiratory Society guidelines.12
For each efficacy endpoint, a total of 15 pair-wise comparisons were performed; superiority testing of all active treatments versus placebo, Foradil® 12µg versus 24µg, FF MDI 19.2µg versus 9.6µg and 7.2µg, FF MDI 9.6µg versus 7.2µg, and non-inferiority testing of all doses of FF MDI versus both doses of Foradil®. PK endpoints measured included additional area-under-the-curve analyses and calculation of the maximum observed plasma concentration (Cmax).
Determination of Sample Size
Calculations to determine sample size were based on the primary endpoint, FEV1 AUC0-12. In order todetect a difference of 100mL in superiority testing between FF MDI and placebo, and to provide adequate power to conduct PK comparisons between FF MDI 9.6µg and Foradil® 12µg, a sample size of 48 patients was chosen.
Statistical Analysis
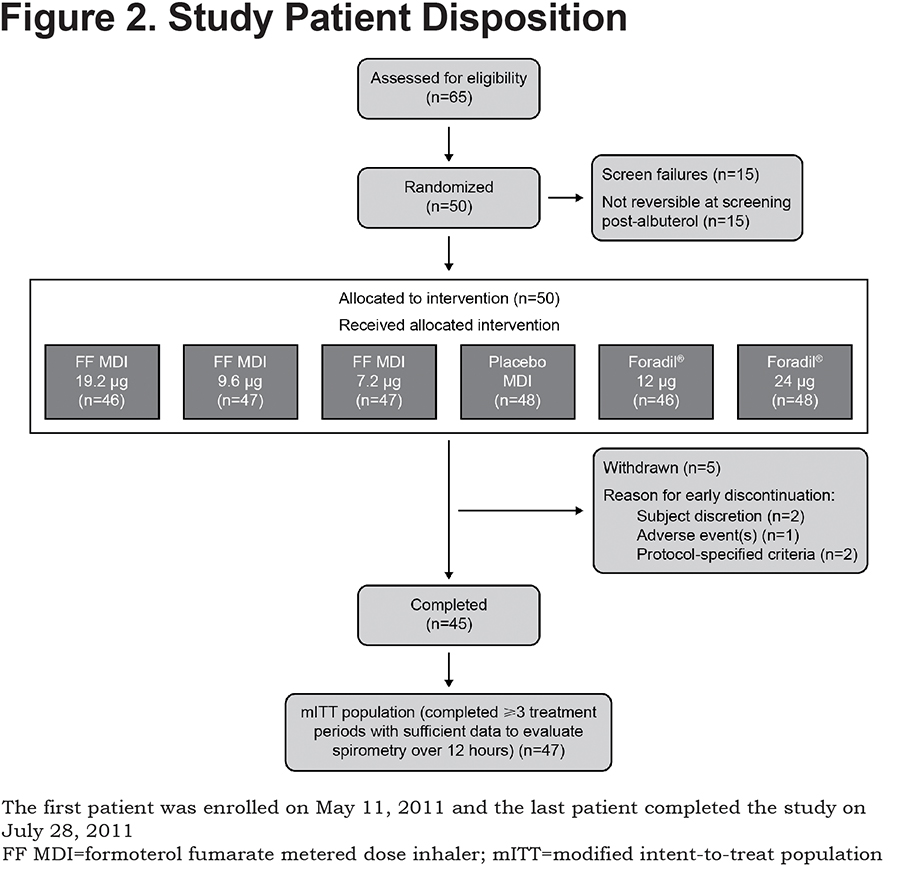
The modified intent-to-treat (mITT) population was considered the primary population for efficacy analyses. This included all intent-to-treat patients who completed at least 3 treatment visits with sufficient data to permit evaluation of spirometry over 12 hours and who did not meet certain pre-specified exclusionary protocol deviations. These patients were included in the PK-evaluable population following evaluation for protocol deviations that may have impacted on PK endpoints. The safety population included all patients who received at least 1study treatment and completed the subsequent post-dose safety assessment.
Baseline demographic data and safety data were summarized descriptively. A linear mixed-effects model was used to compare FF MDI to placebo, with FEV1 AUC0-12 as the dependent variable, and baseline, period and sequence as fixed effects and subject as a random effect. In FEV1-based efficacy analyses, baseline FEV1 was defined as the average of the pre-dose values measured during that study period. The primary efficacy analysis involved 3 treatment comparisons for superiority, using hierarchical testing to control the family-wise Type I error. No claims of superiority over placebo MDI were made for a given FF MDI dosage unless the higher dosage-level showed superiority to placebo MDI. The least squares mean (LSM) and the standard error of the mean (SEM) with the corresponding 2-sided 95% confidence interval (CI) were provided for each treatment based on the model. The LSM difference between each 2 treatments and the SEM, as well as the corresponding 2-sided 95% CIs, were also provided based on the model. A similar method was used to perform non-inferiority comparisons of FF MDI to Foradil® 12µg where a margin of 100mL was pre-specified.
Descriptive statistics and analysis of variance (ANOVA) were used to analyze PK data. Plasma FF PK parameters were calculated using a non-compartmental approach based on the individual plasma concentration profiles over the 0–12 hour time interval using actual sampling time. An ANOVA was performed based on ln-transformed PK parameters (AUC0-12 and Cmax) of FF, with and without dose-normalization using a mixed-effect model, with treatment and period as fixed effects, and patient as a random effect. The ln-transformed results were back-transformed to the original scale by exponentiation to obtain geometric LSM for each treatment and the ratio of PK parameters was summarized using descriptive statistics.
Results
Baseline Characteristics
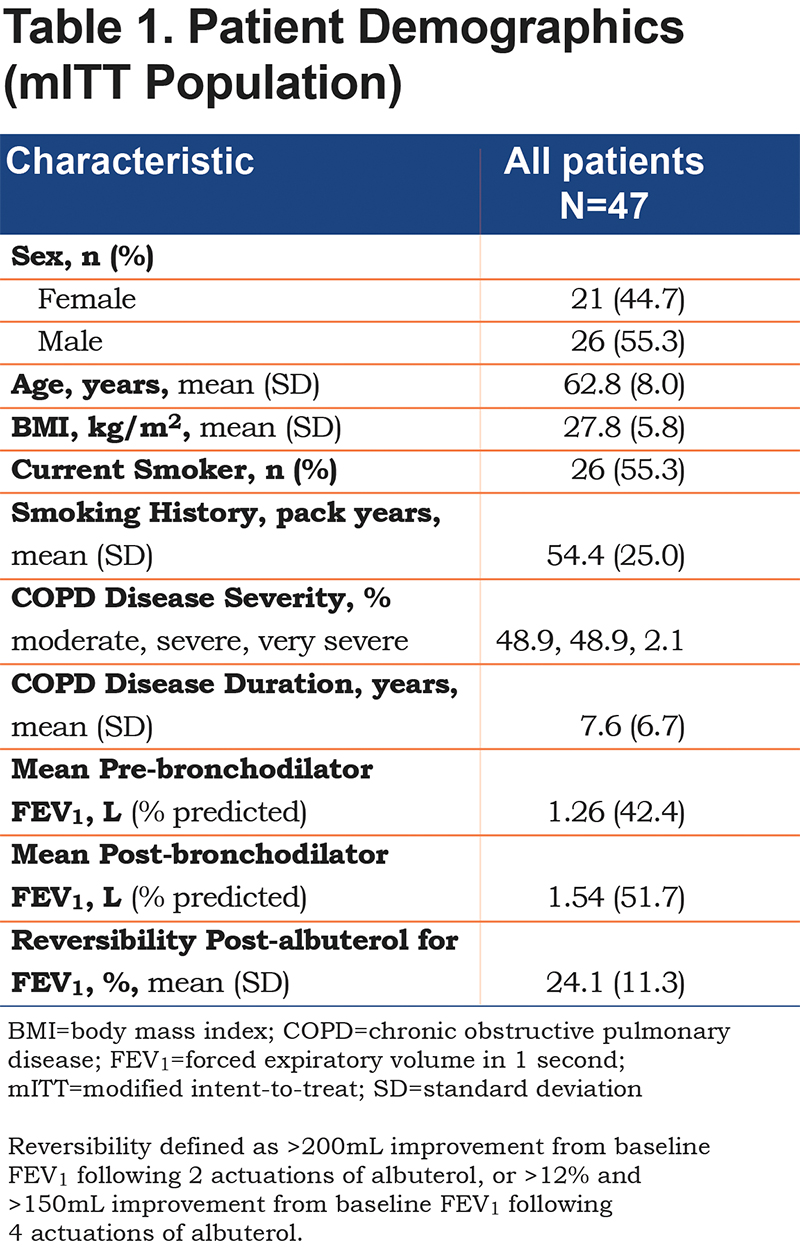
A total of 65 patients underwent screening and, of these, 50 were randomized to study treatment sequences and 45 completed the study (Figure 2). Baseline demographics of the mITT study population (≥3 treatments completed and sufficient data to evaluate spirometry over 12 hours [n=47]) are presented in Table 1. The mean (standard deviation [SD]) age of the mITT population was 62.8 (8) years and the study included more male than female patients (55.3% versus 44.7%, respectively). Smoking history, expressed in terms of pack years (SD), was 54.4 (25.0). At screening, the mean FEV1 was 1.26 L pre-dose (42.4% of predicted) and 1.54 L post-dose (51.7% of predicted). All patients in the mITT population demonstrated reversibility post-albuterol administration and the mean percentage improvement in FEV1 post-albuterol was 24.1%.
Primary Efficacy Endpoint
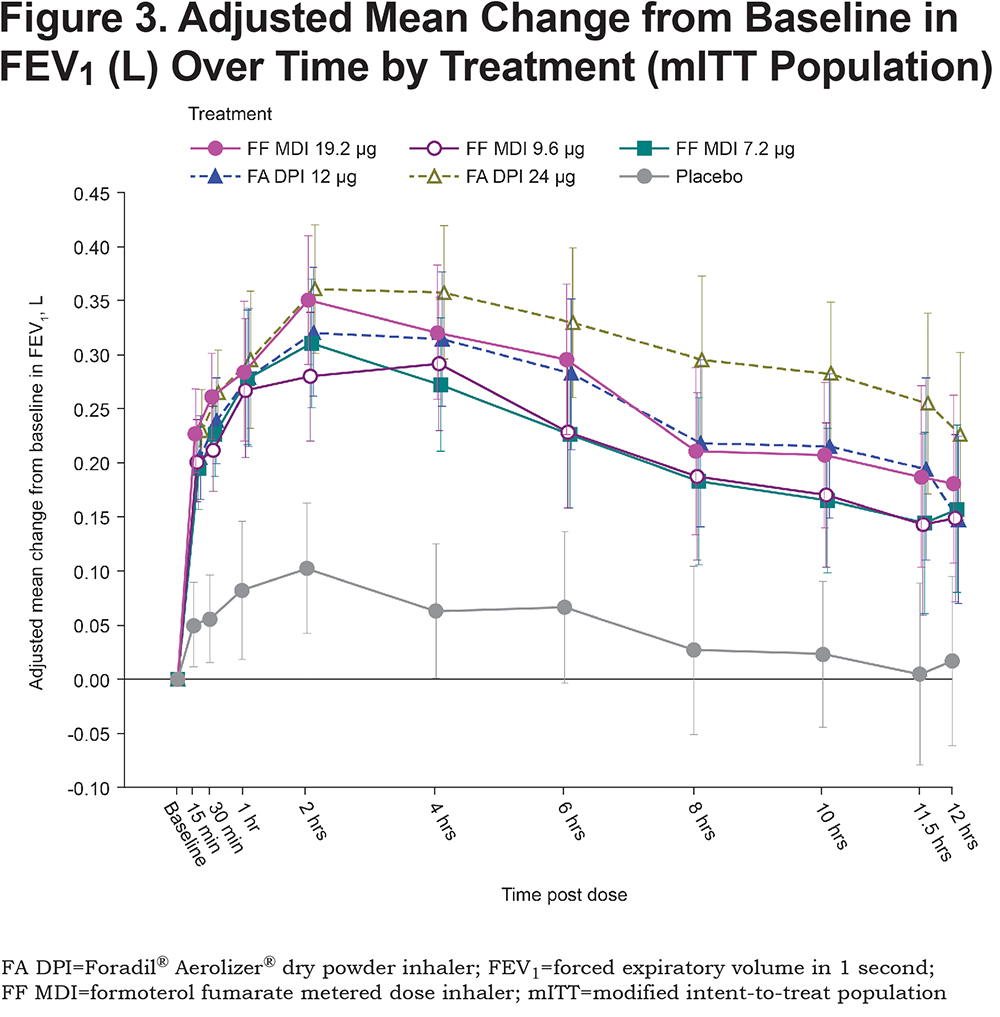
All active treatments were superior to placebo MDI (p<0.0001) in terms of FEV1 AUC0-12, relative to baseline (Figure 3). The differences between each of the active treatments, FF MDI (19.2, 9.6 and 7.2μg) and Foradil® (12 and 24μg), and placebo MDI exceeded the pre-defined clinically relevant effect size of 100mL for FEV1 (adjusted mean differences: 164 to 260mL). Compared with placebo MDI, improvement in FEV1 AUC0-12 (adjusted mean [SE]) following administration of FF MDI was greatest with the 19.2µg dose (211 [17.2]mL, 169 [17.2]mL and 164 [17.1]mL for 19.2, 9.6 and 7.2μg, respectively). For the open-label comparator, improvement in lung function with Foradil® relative to placebo MDI was also dose-dependent and was greatest for the 24µg dose (260 [17.1]mL and 208 [17.3]mL for 24 and 12µg, respectively).
Secondary Efficacy Endpoints
Within the 3 doses of FF MDI, the 19.2µg dose was superior in lung function improvement, compared with each of the lower doses, 9.6 and 7.2μg (adjusted mean difference [95% CI] = 42 [9, 76]mL, p=0.0129; and 47 [14, 80]mL, p=0.0052, respectively). The same dose-dependent superiority for bronchodilation was observed for the higher 24μg dose of Foradil®, compared with the 12μg dose (difference [95% CI] = 52 [19, 85]mL, p=0.0022). These differences for the high and low dose comparisons support the assay sensitivity of this study and thus allow conclusions about non-inferiority to be drawn.
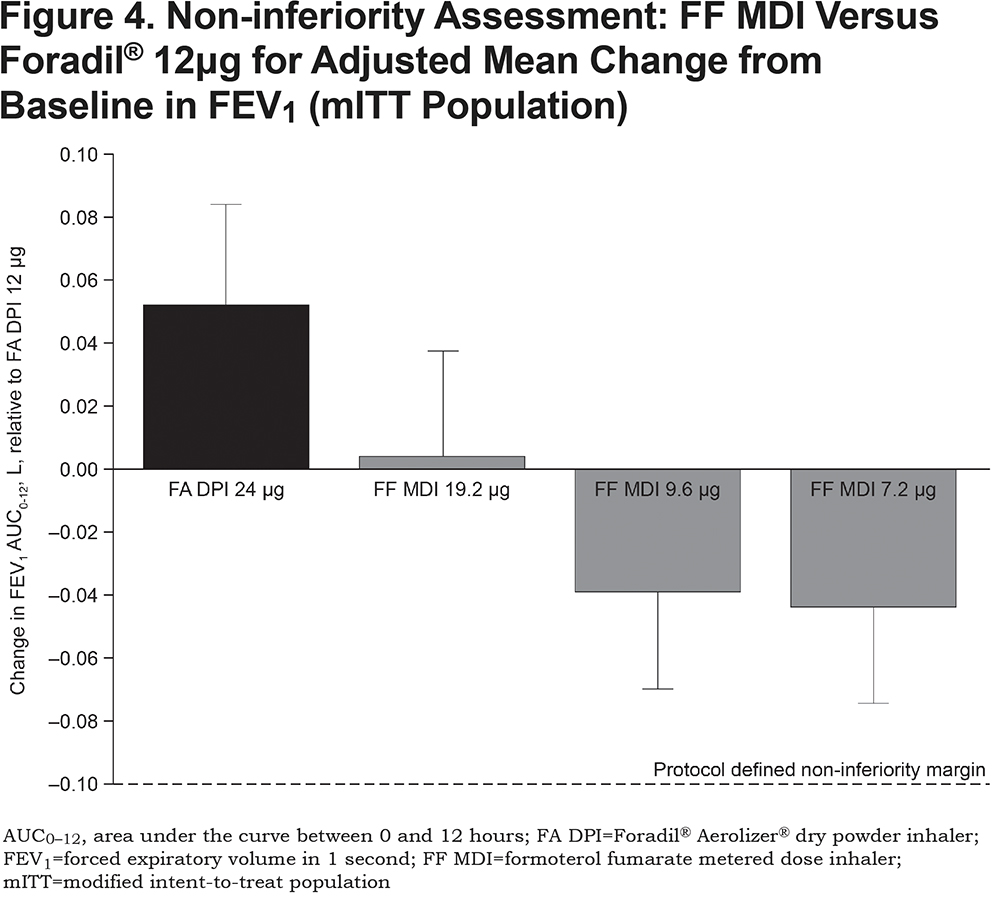
For all doses of FF MDI, the change in FEV1 AUC0-12 over the 12-hour post-dose period demonstrated non-inferiority to Foradil® 12μg (difference [95% CI] = 4 [-30, 37]mL; -39 [-72, -5]mL; and -44 [-77, -11]mL, for 19.2, 9.6 and 7.2μg respectively; Figure 4), according to the protocol-defined non-inferiority margin of 100mL. The highest 19.2μg dose of FF MDI also demonstrated non-inferiority to Foradil® 24μg (difference [95% CI]: -48 [-81, -15]mL).
The adjusted mean peak change from baseline in FEV1 was superior to that associated with placebo MDI for all active treatments (p<0.0001; Table 2). For both active treatments, the greatest changes were associated with the highest doses (403 and 433mL for FF MDI 19.2μg and open-label Foradil® 24μg, respectively). Similarly, all active treatments showed superiority to placebo MDI in terms of the change from baseline at 12-hour post-dose trough FEV1 (p<0.0001; Table 2). The adjusted mean changes from baseline exceeded the clinically relevant effect size of 100mL for FEV1 (adjusted mean differences: 143 to 243mL).
Systemic Exposure
Mean concentration-time profiles of FF following single-dose administration of FF MDI (19.2, 9.6 and 7.2μg) and Foradil® (12 and 24μg), are presented in Figure 5. An approximate 2-fold increase in exposure to FF (in terms of Cmax and AUC0-12) was observed following single-dose administration of FF MDI 19.2μg compared with FF MDI 9.6μg. A similar trend was apparent for differences in exposure to FF between FF MDI 9.6μg and FF MDI 7.2μg (1.3-fold increase). These observations suggest a linear increase in the PK of FF within the dose range tested.
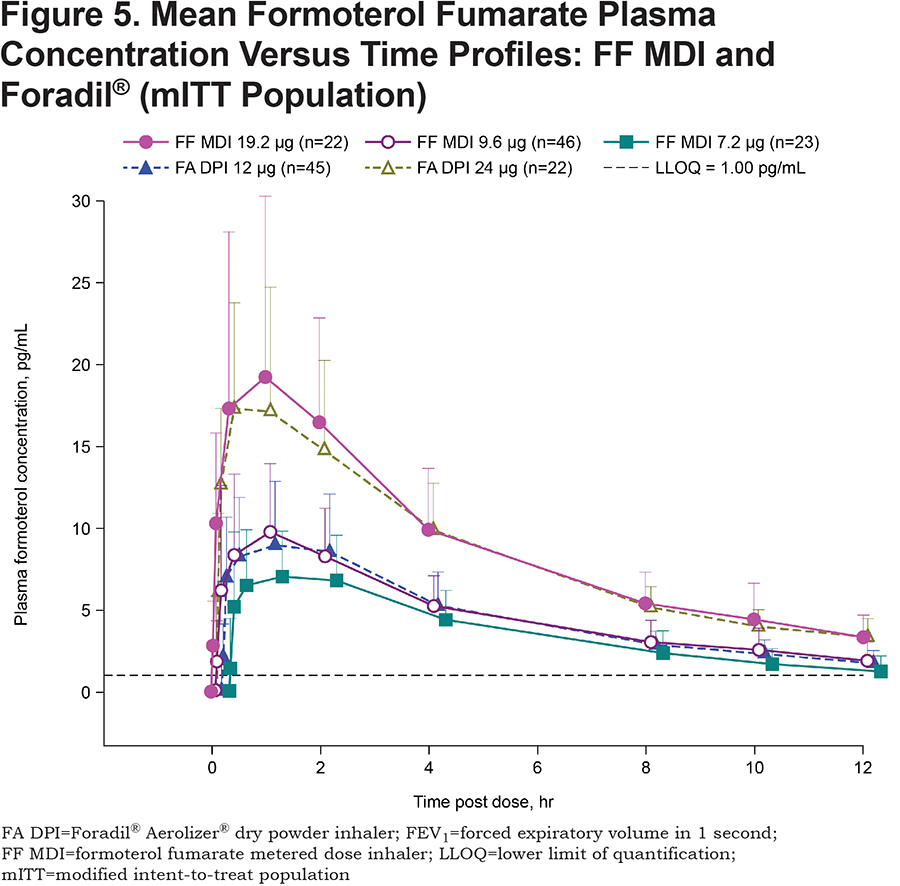
Exposure to FF (Cmax and AUC0-12) was comparable between FF MDI 9.6μg and Foradil® 12μg such that the ratios of the adjusted geometric mean (90% CI) for ln-transformed Cmax and AUC0-12 were 95.5 (86.1, 106.0) and 95.2 (86.7, 104.5), respectively. Given that the 90% CIs for these ratios were within the 80%–125% bioequivalence limits, equivalent systemic exposure for FF MDI 9.6μg and Foradil® 12μg can be concluded. The route of absorption (pulmonary, oropharyngeal, or gastrointestinal) of FF MDI 9.6μg and Foradil® 12μg was not investigated, so pulmonary bioavailability data are not available.
Safety and Tolerability
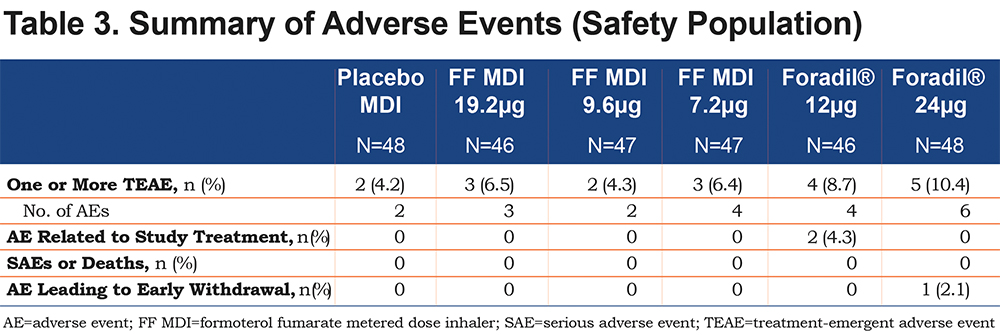
A total of 8 participants experienced at least one adverse event after administration of FF MDI (7.2µg: 6.4%, 9.6µg: 4.3%, 19.2µg: 6.5%) compared with a total of 9 patients following administration of Foradil® (12µg: 8.7%, 24µg: 10.4%) (Table 3). Tremor and nasopharyngitis were the most frequently reported adverse events, each reported in 2 (4.0%) patients overall; tremor was reported in 1 patient while receiving FF MDI 19.2μg and again when receiving Foradil® 12μg, and in another patient while receiving Foradil® 12μg, and nasopharyngitis was reported in 2 patients receiving Foradil® 24μg. No deaths or serious adverse events were reported during the study. One patient discontinued early from the study during treatment with Foradil® 24µg due to an increase in hepatic enzymes, an adverse event considered unrelated to treatment.
Discussion
The current study demonstrated that FF 9.6µg formulated using novel Co-Suspension delivery technology and inhaled via MDI provides non-inferior improvements in lung function and is bioequivalent to Foradil® 12μg in patients with moderate-to-severe COPD, who showed reversibility to the short-acting beta2-agonist, albuterol. All doses of FF MDI (7.2, 9.6 and 19.2µg) showed superior bronchodilator efficacy compared with placebo MDI.
This Phase II study is one of a number of dose-ranging studies undertaken with FF MDI. Additional dose-ranging studies have been performed using the long-acting muscarinic antagonist (LAMA) glycopyrrolate also formulated using Co-Suspension delivery technology for inhaled delivery via the MDI. The overall aim is to widen the treatment options for patients with COPD by offering the convenience of combining multiple active agents within a single inhaler, which are aligned with treatment guidelines for the management of COPD.13 Thus, establishing the effective dose range for the individual components using this delivery system provides the foundation for the investigation of both dual and triple inhaled fixed-dose combinations incorporating Co-Suspension delivery technology.
FF MDI 9.6µg showed non-inferior lung function improvement compared with Foradil® 12µg. The 100mL margin for non-inferiority was selected on the grounds that it is a change in FEV1 that can be perceived by the patient and has been correlated with fewer relapses following exacerbations.14 In fact, the magnitude of the lower limit of the 95% CI for the difference between Foradil® 12µg and the 9.6µg dose of FF MDI was less than 75mL for all assessments and the point estimates were <40mL. These data demonstrate that FF delivered by MDI is bioequivalent to Foradil®.
Patients were selected for inclusion into the study on the basis of their response to administration of the short-acting beta2-agonist, albuterol. Published studies have shown a greater magnitude of response to bronchodilators in patients with COPD reversible to albuterol, relative to non-reversible patients.15-17 Given this observation, we anticipated that patients who demonstrate reversibility would provide the statistical power necessary to detect differences in lung function improvement between the 2 doses of Foradil® and between the highest doses of FF MDI (9.6μg and 19.2μg). Further studies are necessary to determine whether FF MDI and Foradil® are bioequivalent in the non-reversible COPD population, although smaller bronchodilator responses in these patients may mask potential differences between the 2 compounds.
The study highlighted no major safety concerns, although it is acknowledged that it was not a large sample, participants did not have severe comorbidities and exposure to the study drug was limited. Of the 2 pre-identified adverse events of special interest, tremor and paradoxical bronchospasm, only tremor was reported during the course of the study, following administration of the Foradil® 12µg dose in 2 patients. There were no reports of paradoxical bronchospasm. Reductions in serum potassium levels following administration of FF MDI, a known class effect, were comparable to those observed for Foradil®.
The efficacy, safety and PK outcomes from the current study are consistent with those reported from the first dose-ranging study of FF MDI using novel Co-Suspension delivery technology conducted in patients with moderate-to-severe COPD over the dose range 2.4, 4.8 and 9.6µg9 and also from the second study which explored the efficacy and PK profile of FF MDI 7.2 and 9.6µg administered for 7 days, also in patients with moderate-to-severe COPD.10 In the dose-ranging study, FF MDI 9.6µg was shown to be superior to placebo in terms of FEV1 AUC0-12 and non-inferior to Foradil® 12µg. In the chronic-dosing study, FF MDI 9.6µg had a comparable efficacy profile to Foradil® 12µg. Both studies showed comparable PK profiles between FF MDI 9.6µg and Foradil® 12µg. In this current study, a dose range of FF MDI 7.2, 9.6 and 19.2µg was explored and demonstrated a linear dose response according to bronchodilator effect. The observed PK profile of FF MDI also reflected this linear relationship within the dose range tested. AUC0-12 and Cmax values were approximately doubled following single-dose exposure to FF MDI 19.2µg compared with FF MDI 9.6µg and increased by 1.3-fold following FF MDI 9.6µg compared with FF MDI 7.2µg.
In summary, this study showed that FF MDI 9.6μg bronchodilator response was non-inferior to that of Foradil®12μg, thereby demonstrating bioequivalence. The assay sensitivity achieved within the study confirms the validity of these findings. All doses of FF MDI and Foradil® were well tolerated. This supports the selection of the FF MDI 9.6µg dose for further evaluation in Phase III trials.
Acknowledgements
The authors would like to thank all of the patients and their families, the team of investigators, research nurses and operations staff involved in this study. The authors would also like to thank Everest Clinical Research, who performed the statistical analyses for this study, funded by Pearl Therapeutics, Inc. a member of the AstraZeneca group.Medical writing/editorial support was provided by Catherine Stanton of Complete Medical Communications, funded by Pearl Therapeutics, Inc., a member of the AstraZeneca Group.Co-Suspension is a trademark of the AstraZeneca group of companies. Principal investigators: Charles M. Fogarty, Leonard Dunn, Jack Parrino.
Declaration of Interest
SSethi has received grants from AstraZeneca, Dey, and Pearl Therapeutics, Inc. He has received personal fees from AstraZeneca, Bayer, Boehringer Ingelheim, Cempra, CSL Behring, Forest, GlaxoSmithKline, Merck, Pearl Therapeutics Inc., Pulmonx, Reckitt Benckiser, Sunovion, and Theravance. CF is a consultant and investigator for Pearl Therapeutics, Inc. NAH has served as a consultant and received honoraria from Pearl and Astra Zeneca. His institution has received research grant support on his behalf to conduct clinical trials.
FJM has participated in steering committees on COPD or idiopathic pulmonary fibrosis sponsored by Bayer, Centocor, Forest, Gilead, Janssen, GlaxoSmithKline, Nycomed/Takeda and Promedior. He has participated in advisory boards for COPD or idiopathic pulmonary fibrosis for Actelion, Amgen, AstraZeneca, Boehringer Ingelheim, Carden Jennings, CSA Medical, Ikaria, Forest, Genentech, GlaxoSmithKline, Janssen, Merck, Pearl, Nycomed/Takeda, Pfizer, Roche, Sudler & Hennessey, Veracyte, and Vertex. He has prepared or presented continuing medical presentations on COPD or idiopathic pulmonary fibrosis for the American College of Chest Physicians, the American Thoracic Society, CME Incite, Center for Health Care Education, Inova Health Systems, MedScape, Miller Medical, National Association for Continuing Education, Paradigm, Peer Voice, Projects in Knowledge, Spectrum Health System, St. John’s Hospital, St. Mary’s Hospital, University of Illinois-Chicago, University of Texas Southwestern, University of Virginia, UpToDate, and Wayne State University. FJM has participated in data safety monitoring committees sponsored by GlaxoSmithKline and Stromedix. He has assisted with Food and Drug Administration presentations sponsored by Boehringer Ingelheim, GlaxoSmithKline, and Ikaria. He has spoken on COPD for Bayer, Forest, GlaxoSmithKline, and Nycomed/Takeda. He has participated in advisory teleconferences sponsored by the American Institute for Research, Axon, Grey Healthcare, Johnson & Johnson, and Merion. He has received book royalties from Informa. SR is an employee of AstraZeneca PLC. CO, PD, ES, SStrom, TF, MG, SD and CR are employees of Pearl Therapeutics, Inc.