Running Head: A1PI Therapy and Elastin Degradation in RAPID
Funding Support: CSL Behring, the James P. Mara Center for Lung Disease, the Flight Attendants Medical Research Institute, the Charles A. Mastronardi Foundation, the Ned Doyle Foundation, and the Alpha-1 Foundation all provided funding for this study.
Date of Acceptance: October 3, 2016
Abbreviations: alpha-1 antitrypsin deficiency, AATD; alpha-1 proteinase inhibitor, A1PI; desmosine, DES; isodesmosine, IDES; computed tomography, CT; neutrophil elastase, NE; chronic obstructive pulmonary disease, COPD; total lung capacity, TLC; forced expiratory volume in 1 second, FEV1; functional residual capacity, FRC; 15th percentile CT lung density, PD15; diffusing capacity of the lungs for carbon monoxide, DLCO; body mass index, BMI; standard error, SE
Citation: Ma S, Lin YY, Cantor JO, et al. The effect of alpha-1 proteinase inhibitor on biomarkers of elastin degradation in alpha-1 antitrypsin deficiency: An analysis of the RAPID/RAPID Extension trials. Chronic Obstr Pulm Dis. 2017; 4(1): 34-44. doi: http://doi.org/10.15326/jcopdf.4.1.2016.0156
Introduction
Alpha-1 antitrypsin deficiency (AATD) is a rare genetic disease characterized by low serum levels of alpha-1 proteinase inhibitor (A1PI), also known as alpha-1 antitrypsin.1 The major role of A1PI is to inhibit the proteolytic enzyme neutrophil elastase (NE). NE is released by activated neutrophils and can degrade elastin within connective tissues. Uncontrolled NE causes degradation of lung tissue and, in patients with an absence or deficiency of A1PI, this may lead to the development of emphysema in later life.1 In recent years much has been discovered regarding the broad variability of genetic factors and clinical phenotypes associated with AATD.1 The discovery of AATD and reduced elastase inhibition as a genetic abnormality causing pulmonary emphysema gave a focus to elastin and elastin degradation as significant factors in lung matrix injury and the pathogenesis of pulmonary emphysema.2
Desmosine and isodesmosine (DES/IDES) are 2 crosslinking amino acids which occur only in the mature elastin fiber. Upon elastin degradation they are released and circulate into the plasma, urine and sputum, where they can be measured with sensitivity and accuracy.3-5 Levels of DES/IDES are elevated in chronic obstructive pulmonary disease (COPD), both related and not related to AATD, and have become useful biomarkers of the presence of COPD and its responses to therapy.5-7 Preliminary evidence also suggested that plasma DES levels may be altered in patients with AATD,8 and later studies confirmed that plasma levels of DES may be a suitable long-term biomarker for use in AATD.9 More recently it has been demonstrated that A1PI therapy in AATD consistently reduced levels of DES/IDES in plasma and bronchoalveolar lavage fluid, with A1PI therapy administrated either intravenously or by aerosol.10
The RAPID trial (NCT00261833; N=180) was a multi-center, double-blind, randomized, placebo-controlled trial of A1PI treatment in patients with AATD conducted over 2 years, with an extension period of 2 more years (RAPID Extension trial, NCT00670007; N=140).11 Primary endpoints were computed tomography (CT) measurements of lung density at total lung capacity (TLC) and functional residual capacity (FRC) combined and the 2 separately,11 as CT is considered a more valid measurement of efficacy than spirometric parameters.12 RAPID showed a statistically significant reduction in the rate of lung density decline in patients receiving A1PI compared with placebo (annual rate of lung density loss at TLC was -1.45 g/L per year versus -2.19 g/L per year, respectively; p=0.03), indicating that A1PI is able to preserve lung tissue density and reduce emphysema progression.11 In the RAPID Extension study, patients previously on placebo switched to A1PI therapy (Delayed-Start group) and subsequently demonstrated reduced lung density decline; however, these patients did not catch up with those on A1PI throughout RAPID (Early-Start group), indicating that A1PI therapy is disease-modifying.11 Prior studies using CT lung density as an endpoint in patients with AATD receiving A1PI therapy for shorter periods of time and with fewer participants suggested similar efficacy of A1PI therapy in AATD.12-15
The RAPID/RAPID Extension trials, which represent the largest randomized, placebo-controlled trials of A1PI therapy completed to date, provided a unique opportunity to assess biomarkers of body elastin degradation, including pulmonary elastin, over a prolonged period of study (4 years). Our aim was to determine the relationship between the effect of A1PI therapy on DES/IDES and lung density as determined by CT.
Methods
Patients and Study Design
This study was a post-hoc analysis of samples taken from patients during RAPID, a multi-center, double-blind, randomized, parallel-group, placebo-controlled trial. Men and women aged 18–65 years were recruited with emphysema secondary to AATD (with a serum A1PI concentration of <11 μM and a forced expiratory volume in 1 second (FEV1) of 35%–70% of predicted normal; Table 1). The study was conducted in 28 study centers in 13 countries. Participants were excluded if they had smoked tobacco within 6 months before recruitment or had undergone or were on the waiting list to undergo lung transplantation, lobectomy, or lung volume reduction surgery; or had selective immunoglobulin A deficiency. Concurrent A1PI treatment was not allowed. Patients provided written informed consent and researchers obtained approval from local institutional review boards. Patients’ plasma samples were stored at -20oC until analyzed.
Randomization and Masking
Randomization was conducted centrally 1:1 with patients who completed a screening period of 1-4 weeks of treatment with A1PI or matching placebo, as previously described.11 The post-hoc measurement of DES/IDES levels reported here was conducted using randomized plasma samples, i.e., without knowledge of treatment assignment.
Procedures
Randomized patients received either A1PI at 60 mg/kg per week (N=93; Zemaira®/Respreeza®; CSL Behring, Pennsylvania) or placebo (N=87) of equal volume intravenously for 24 months during RAPID. Patients completing the double-blind portion (in both the A1PI and placebo groups) of the protocol were eligible to receive open-label treatment with A1PI 60 mg/kg per week for a further 2 years in the RAPID Extension trial (only non-U.S. patients were enrolled because of unavailability of A1PI treatment in the non-U.S. countries).
Pulmonary Analyses
As part of RAPID/RAPID Extension, spiral CT scans were performed at TLC and FRC.12 Lung density measurements in Hounsfield units were transformed to g/L and a physiological volume correction was applied to the 15th percentile CT lung density (PD15), as described previously.13 Lung exacerbations were defined according to the Anthonisen criteria.16
Desmosine and Isodesmosine Analysis
All participants from the intention-to-treat population from RAPID/RAPID Extension were eligible for the DES/IDES analysis. The numbers reported reflect the number of evaluable samples (some loss occurred due to insufficient sample volume, withdrawals, etc.). Plasma samples were acid-hydrolyzed for 24 hours in 6 N HCl at 110oC and were then applied to a CF1 cartridge to purify. A synthetic desmosine-d4 served as the internal standard for processing plasma samples and measuring DES and IDES.17 High-performance liquid chromatography and tandem mass spectrometry methods were used as previously described.17 Details of the methods can be found in the online supplement. Analyses of DES/IDES levels were performed in triplicate; the coefficient of variation of the method is 7%.
Statistical Analysis
Analysis of covariance was used to compare DES/IDES values at months 3, 12, 24, 36, and 48 to baseline values and between treatment groups at each time point. Spearman correlations were used to evaluate changes in the levels of DES/IDES versus the following: CT lung density decline at TLC at month 24; FEV1 at baseline and months 24 and 48; and the diffusing capacity of the lungs for carbon monoxide (DLco) at baseline and month 24. A one-tailed t-test was used to evaluate differences between body mass index (BMI) groups, since obesity has been associated with inflammatory biomarkers and degeneration of body elastin18,19 and only one direction of analysis was of interest.
Results
Patient Characteristics
The baseline characteristics of patients in the placebo and A1PI groups are summarized in Table 1. The 2 groups were well matched. Baseline values for age, sex, FEV1 % predicted, CT lung density and A1PI serum levels in each group were very similar to those reported previously in each group in the full RAPID study population.11
Changes From Baseline DES/IDES Levels Through 48 Months
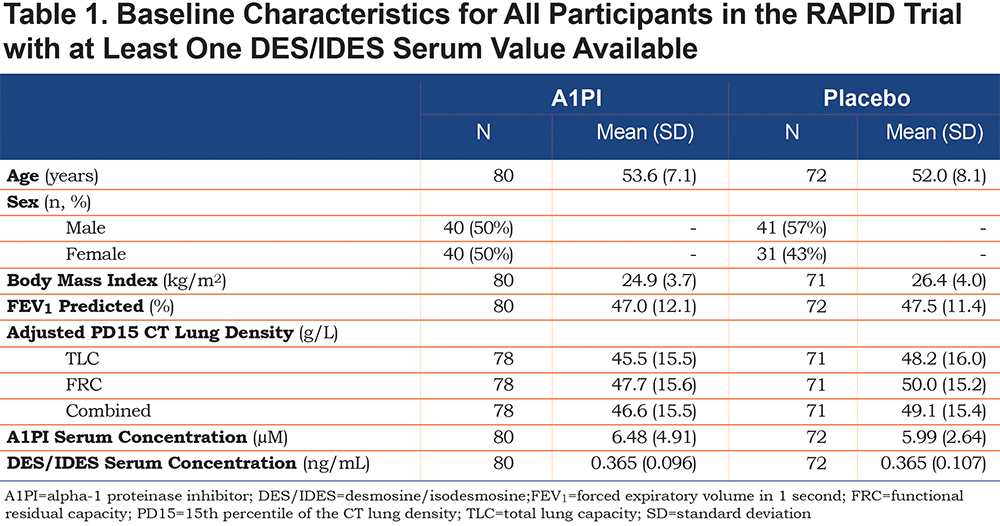
During RAPID, significant decreases in DES/IDES levels (mean ± SE) from baseline were observed in patients receiving A1PI at all time points: month 3 (-0.013; 95% CI -0.024, -0.002; p=0.024), month 12 (-0.031; 95% CI -0.040, -0.021; p<0.001), and month 24 (-0.036; 95% CI -0.048, -0.023; p<0.001) (Figure 1a). Further decreases in DES/IDES were observed during RAPID Extension (Early-Start group) in patients continuing to receive A1PI. A small increase in DES/IDES levels from baseline was observed with placebo-treated patients in the RAPID study (Delayed-Start group) at months 3 and 12; although these were not significant, a significant increase was observed at month 24 (n=54; 0.016; p=0.018). A significant treatment difference between groups was observed at months 3 (-0.021; 95% CI -0.037, -0.004; p=0.026), 12 (-0.040; 95% CI -0.055, -0.025; p<0.001), and 24 (-0.052; 95% CI -0.070, -0.034; p<0.001) during the RAPID study. Upon treatment with A1PI, the Delayed-Start group showed a significant reduction in DES/IDES levels from baseline; month 36 (-0.065; 95% CI -0.082, -0.049; p<0.001) and month 48 (-0.074; 95% CI -0.092, -0.057; p<0.001).
Since multiple measurements of participants’ biomarker levels were performed in each study arm, repeated ANOVA methodology was applied post-hoc and the overall comparison between treatment arms was also statistically significant (p<0.0001). This analysis showed lower DES/IDES levels for participants on treatment, with differences (treatment − placebo) that were statistically significant at month 12 (-0.030; 95% CI -0.060, -0.002; p=0.039), month 24 (-0.044; 95% CI -0.073, -0.016; p=0.003), and month 48 (-0.032; 95% CI -0.055, 0.003; p=0.032). Values at baseline (0.008; 95% CI -0.020, 0.037; p=0.564) were similar, and although point estimates at month 3 (-0.011; 95% CI -0.040, 0.017; p=0.437) and month 36 (-0.026; 95% CI -0.055, 0.003; p=0.080) suggested lower DES/IDES levels on treatment, these differences were not statistically significant.
Correlation of DES/IDES with CT Lung Density and Serum A1PI Concentration
Levels of DES/IDES (change from baseline) showed a weak but significant inverse correlation with CT scan lung density decline (n=120; R=-0.256, p=0.005) at month 24 (Figure 1b). Decreased rates of lung density decline (g/L/year) were associated with reductions in plasma DES/IDES levels (ng/mL) (Figure 1b). Significant correlations were shown previously between the nadir levels of A1PI and the preservation of lung density by CT.11 We show similar significant correlations between nadir levels of A1PI and reductions in plasma DES/IDES at month 24 (n=119; R=-0.460 and p<0.001) (Figure 1c), with higher serum A1PI levels associated with reductions in plasma DES/IDES levels (ng/mL).
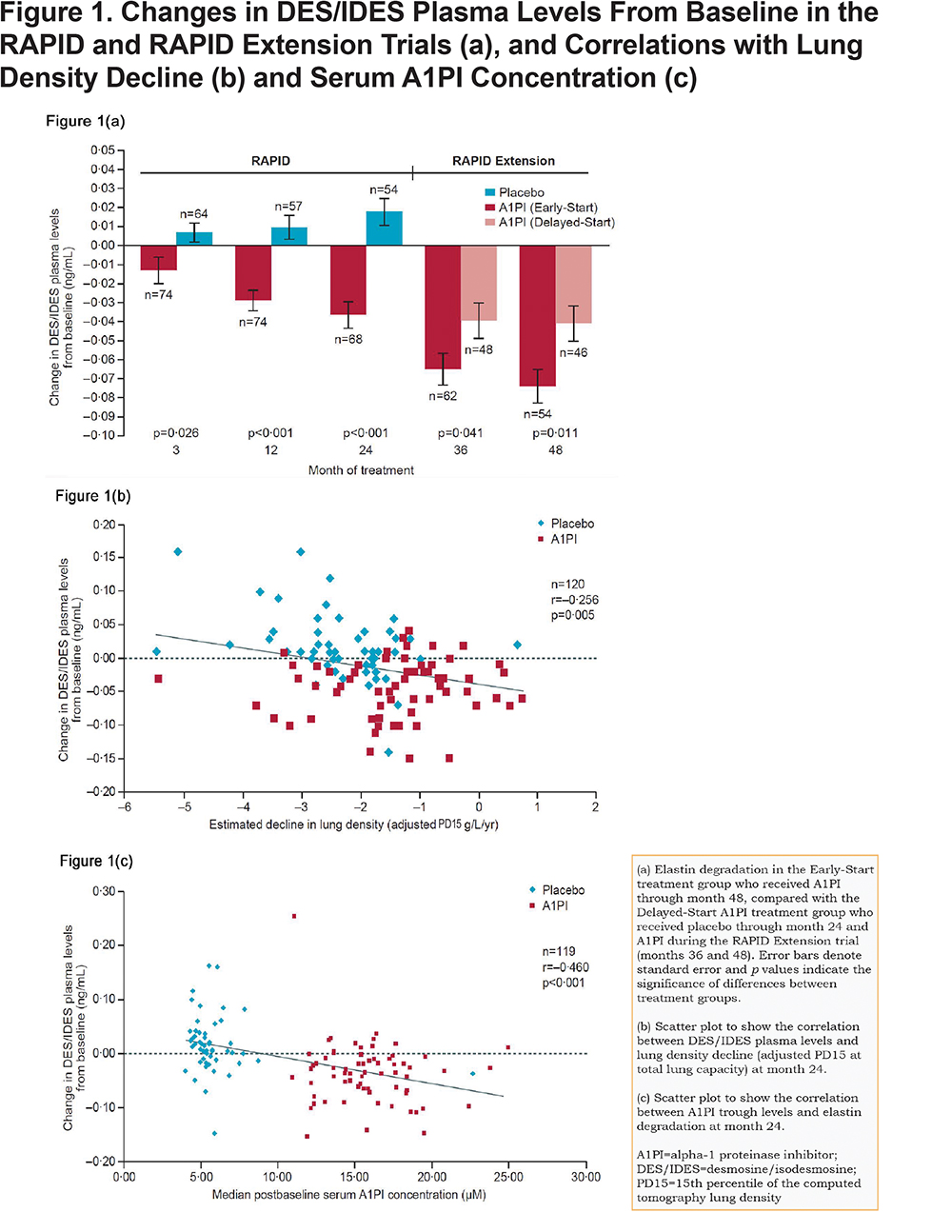
Correlation of DES/IDES with FEV1 and DLCO
Plasma levels of DES/IDES were weakly but significantly correlated with FEV1 and DLCO (Figures 2a-e). Significant correlations to FEV1 were found at baseline (n=152; R=-0.197 and p=0.015), month 24 (n=136; R=-0.241 and p=0.005), and month 48 (n=114; R=-0.204 and p=0.03) (Figures 2a-c). Similarly, significant correlations to DLCO were found at baseline (n=152; R=-0.284 and p<0.001) and month 24 (n=136; R=-0.236 and p=0.006) (Figures 2d-e). Reduction in pulmonary parameters, FEV1 and DLCO, was associated with increasing levels of plasma DES/IDES (ng/mL) at all time points. Correlation data for DLCO was not available for month 48 as DLCO was not assessed during the RAPID Extension trial.
Exacerbations
Thirty-nine patients experienced their first exacerbation within the first 2 months of the study and their DES/IDES level (mean ± SE) was 0.37 ± 0.02 ng/mL. Twenty-five patients had their first exacerbation between 2 and 6 months, with a DES/IDES level (mean ± SE) of 0.36 ± 0.02 ng/mL. Eleven patients had their first exacerbation between 1.5–2 years; the mean ± SE DES/IDES level in this group was 0.35 ± 0.03 ng/mL. Although DES/IDES levels decreased slightly as time to first exacerbation increased, the differences at each time point were not statistically significant. A Poisson regression model was fitted to the data (p<0.0001) and showed that for every 0.1 ng/mL increase in baseline DES/IDES levels, the average exacerbation rate increased by 35% over 2 years. Treatment arm was not significant in the model.
DES/IDES and Body Mass Index
In RAPID, individuals with a high BMI received higher weight-related doses of A1PI therapy, which in turn produced a greater effect on lung density preservation, as reported previously.11 We examined the effect of BMI on DES/IDES levels in this same cohort. At baseline, before beginning A1PI therapy, participants with BMI <30 kg/m2 (n=138) had mean ± SE DES/IDES levels of 0.36 ± 0.01 ng/mL, while participants with BMI ≥30 kg/m2 (n=14) had DES/IDES levels of 0.42 ± 0.04 ng/mL (p=0.023, one-tailed t-test). Despite these differences, baseline BMI did not affect the impact of A1PI therapy on DES/IDES levels. At subsequent time points, DES/IDES values (ng/mL) in participants with low versus high BMI were, respectively: month 12, 0.34 ± 0.01 (n=121) versus 0.37 ± 0.03 (n=20); month 24, 0.34 ± 0.01 (n=117) versus 0.38 ± 0.04 (n=18); month 48, 0.29 ± 0.01 (n=98) versus 0.33 ± 0.02 (n=14). One-tailed t-test p values were significant at month 48 (p=0.033). The high BMI cohort showed significant reductions in mean DES/IDES levels with A1PI therapy, from 0.40 ± 0.05 ng/mL at baseline to 0.32 ± 0.03 ng/mL at month 48. No statistically significant difference was observed regarding the effect of A1PI therapy on the lowering of plasma DES/IDES in the high versus the low BMI cohort.
Discussion
The results presented here demonstrate for the first time a correlation between biomarkers of elastin degradation and a clinical marker of emphysema progression (CT lung density) in patients with AATD. Our study measured the levels of biomarkers of elastin degradation, DES/IDES, in plasma in a population of patients with AATD receiving or not receiving A1PI therapy during the 2-year RAPID trial and also in those continuing A1PI therapy (Early-Start) or switching to A1PI therapy (Delayed-Start) for the additional 2 years of the RAPID Extension trial. We found that biomarker levels in patients receiving A1PI treatment decreased throughout RAPID and continued to decrease during the 2-year extension trial (months 36 and 48). Significant differences in the change from baseline DES/IDES levels were found between placebo and treated groups as early as 3 months after initiating A1PI therapy (p=0.026). This finding is consistent with the mechanism of action of A1PI – increasing the serum level of A1PI reduces the breakdown of elastin in the lung by NE and reduces the levels of DES/IDES. Greater differences were found at months 24 and 48, suggesting that the positive effects of A1PI therapy may be cumulative and occur to a greater degree with advancing time. Beneficial changes in elastin-elastase interactions may occur with prolonged A1PI therapy by mechanisms which are currently unknown. In addition, a small increase in the change in DES/IDES levels from baseline was observed in the placebo (Delayed-Start) group at month 24 during the RAPID trial. The change in biomarker levels from baseline throughout placebo treatment is consistent with disease progression and loss of lung tissue density caused by uncontrolled breakdown of elastin by NE.
A weak but significant (p=0.005) correlation between change in DES/IDES levels and CT lung density decline was observed. Since elastases circulate in blood it is likely that the levels of DES/IDES measured in plasma reflect the status of elastin degradation in the whole body, including the blood vessels, skin, and lungs. Because the structural stability of lung elastin is necessary to maintain alveolar structure, it would be reasonable to conclude that the preservation of lung density as determined by CT is consistent with a reduced level of elastin breakdown in the lung per se. This result is also in agreement with previous findings of reduced DES/IDES in bronchoalveolar lavage fluid from patients with AATD receiving A1PI therapy.10 However, it remains possible that A1PI therapy increases inhibition of elastases which affect elastin degradation in organ systems other than the lung and which also contribute to total DES/IDES plasma levels. Other organ systems undergoing elastin degradation include the coronary circulation as well as the systemic arterial system, where there is evidence of a different pattern of coronary atherosclerosis in COPD, as well as increases in arterial blood flow rate consistent with a stiffening of arterial walls in COPD.20 Such possible effects in AATD-related COPD deserve investigation. During the RAPID and RAPID Extension studies no evidence of white blood cell (neutrophil) elevation or systemic inflammation was observed. It can therefore be considered that any breakdown of elastin in the systemic circulation caused by release of NE from activated neutrophils was not related to episodes of infection and leukocytosis in the study.
Weak but significant correlations to changes in DES/IDES levels were also observed with pulmonary function measures. DES/IDES plasma levels correlated significantly with both FEV1 (p=0.015; p=0.005; p=0.029) and DLco (p<0.01; p=0.006), demonstrating a correlation between biochemical markers and clinical markers of lung disease progression. A correlation was also observed between nadir levels of A1PI and levels of DES/IDES; higher post-baseline serum levels of A1PI were associated with reductions in DES/IDES levels. This supports the use of A1PI therapy to increase A1PI serum levels and reduce the breakdown of elastin and preserve lung tissue. Furthermore, reductions in DES/IDES levels with higher A1PI serum levels indicate that the therapeutic effect is related to the dose of A1PI and higher doses may be associated with further reductions in elastin degradation toward normal levels of plasma DES/IDES.
Of interest in the cohort we studied are the plasma levels of DES/IDES in obese participants. Studies have demonstrated an increase in inflammatory biomarkers in high BMI individuals.18 Increased inflammatory state in obesity may promote increased degeneration of body elastin, including vascular elastin, predisposing individuals to vascular disease which is recognized to be increased in obesity.19 However, these results should be noted with caution since higher BMI levels may confound CT densitometry analysis.
Results from this study provide support for the use of biomarkers of elastin degradation to monitor emphysema progression and treatment response in patients with AATD and, going forward, potentially in patients with non-AATD COPD as well. Biomarker levels could be used in clinical practice to assess therapeutic efficacy in individual patients using the range of published normal values as baseline: 0.19 ± 0.01 ng/mL5; 0.21 (0.19–0.24) ng/mL.7 Changes in DES/IDES observed in our study appear small (within 0.1 ng/mL), necessitating the use of sensitive analytical equipment; however, this may be due in part to the relatively low dose of A1PI used in these studies. During RAPID/RAPID Extension, patients received only a single dose, 60 mg/kg per week, of A1PI11; however, the effect of higher A1PI doses has been recently reported. A recent study reported that a dose of 120 mg/kg provided more favorable physiologic A1PI plasma levels than a dose of 60 mg/kg,21 and may also cause a larger change in DES/IDES levels.
Although changes in DES/IDES were small, measurements were stable and showed low variability both within and between individual patients. The observed changes in DES/IDES are expected to be clinically relevant as the level of DES/IDES in healthy individuals is low.5 However, DES/IDES levels, as biomarkers, react to clinical events such as respiratory exacerbations, as previously shown in COPD,7 and therapeutic intervention, as shown here. Our study also suggested that increasing exacerbation rate may be associated with increasing DES/IDES levels. The changes we observed in DES/IDES levels from baseline to month 24 represent a 22% decrease in the Early-Start group and a 14% decrease in the Delayed-Start group. Furthermore, changes in DES/IDES correlated weakly, although significantly, with CT lung density (p<0.05), which is an anatomical index of disease in the lung and a chemical marker of a matrix structural constituent of the lung. Changes in DES/IDES were also correlated weakly but significantly with other clinical markers of lung disease progression, such as FEV1 and DLCO, which are commonly used to monitor emphysema progression.
Our findings are consistent with previously published studies indicating a potential association between elastin biomarker levels and COPD and/or AATD. Previous studies have found DES/IDES levels to be altered in COPD and AATD patients6-8 and elastin biomarkers have been shown to correlate with pulmonary measures21 and emphysema progression,7,22 indicating their potential utility as a long-term biomarker for use in AATD.
In a recent study of patients from the Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) study, Ongay et al23 demonstrated a significant difference in urinary DES between patients with COPD and healthy individuals (p=0.007), after adjusting for confounding factors including smoking habits, gender, age and BMI. However, the authors concluded that a single measurement of urinary DES excretion could not predict lung function decline in individuals with stable COPD, due to the impact of these confounding factors and low assay sensitivity and specificity. Another study of patients from the ECLIPSE trial by Rabinovich et al24 indicated that plasma DES levels were not correlated to emphysema or disease progression, although they were associated with mortality. However, this study was limited by the fact that measurements between the 2 cohorts were different and a lack of repeated sampling in the second cohort meant that outcomes relating to the relationship between plasma DES and emphysema could not be validated .24 Furthermore, patients included within the ECLIPSE study represent a different population to that reported herein. Whereas COPD is associated with emphysema plus a range of symptoms including chronic bronchitis and diseases of the airways, AATD is more directly associated with emphysema due to unchecked degradation of lung tissue by NE. Also, the progression of COPD is notoriously variable, with some patients experiencing a stable course while others suffer relentless progression with frequent acute exacerbations.25 Therefore, it is possible that biomarkers such as DES/IDES might be more suitable for studying disease progression in AATD than in COPD.
The potential limitations of our study should be mentioned. Although intra- and inter-patient variations in DES/IDES seen in this study were small, diet has been implicated in changes in DES/IDES,26 which was not accounted for. Also, changes in DES/IDES could potentially reflect degradation of elastin in other body tissues, such as the skin.27 In addition, our study included samples from a group composed of 50% males in the Early-Start group, but 57% of males in the Delayed-Start group; however, these percentages are both close to the overall mean and considering the sample size of this study the variation is relatively small.
Conclusions
This post-hoc analysis is based on a cohort of patients from RAPID/RAPID Extension, the largest trial completed to date to investigate the effect of A1PI therapy in patients with AATD. Results demonstrate for the first time that DES/IDES levels were significantly reduced versus baseline in patients receiving A1PI from as early as 3 months following therapy (p=0.026). In addition, a correlation between biomarkers of elastin degradation and a clinical marker of emphysema progression (CT lung density) was seen in these patients. These results suggest that therapeutic agents such as A1PI that reduce body elastin degradation can preserve structural stability of the lung over the long-term and should be started early in the course of disease. Results from this study also suggest that DES/IDES levels may be used to evaluate other therapeutic agents which can decrease both elastin degradation and lung matrix injury.
Acknowledgements
This work was supported by funding from CSL Behring, the James P. Mara Center for Lung Disease (New York, New York), the Flight Attendants Medical Research Institute (Miami, Florida), the Charles A. Mastronardi Foundation (Wilmington, Delaware), the Ned Doyle Foundation (New York, New York) and the Alpha-1 Foundation (Miami, Florida), and also by funds from Ethel Kennedy, John Kennedy and Judith Sulzberger (New York, New York).
CSL Behring (King of Prussia, Pennsylvania) funded the RAPID study program and the research work described herein. CSL Behring provided samples from the RAPID and RAPID Extension studies to the corresponding author and had a role in overseeing data analysis. MF, who is an employee of CSL Behring, and JME, who was employed by CSL Behring at the time of the study, participated in data analysis, data interpretation, and writing of the report. Editorial assistance was provided by James Glossop of Meridian HealthComms Ltd, funded by CSL Behring. The corresponding author had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Declaration of Interest
GMT reports grants from CSL Behring during the conduct of the study. JOC has a patent (USPTO 7166437) licensed to MatRx Pharm. KRC reports grants from CSL Behring during the conduct of the study, and grants for other work from AstraZeneca, GlaxoSmithKline, Novartis, Genentech, Grifols, Sanofi, Kamada, Merck, Amgen, Roche and Boehringer Ingelheim. RAS reports grants from CSL Behring during the conduct of the study, and is employed part-time by the Alpha-1 Foundation and AlphaNet, 2 not-for-profit entities that support research and health management in Alpha-1 and that receive part of their funding from pharmaceutical industry sources. NGM reports grants and personal fees from CSL Behring during the conduct of the study. MF is an employee of CSL Behring. JME was employed by CSL Behring at the time of the study. All other authors have nothing to disclose.