Running Head: Aclidinium ASCENT Study Design
Funding Support: The study was initially funded by Forest Laboratories and later funded by AstraZeneca.
Date of Acceptance: August 25, 2017
Abbreviations: chronic obstructive pulmonary disease, COPD; cardiovascular, CV; long-acting muscarinic antagonist, LAMA; Aclidinium Bromide on Long-Term Cardiovascular Safety and COPD Exacerbations in PatieNTs with Moderate to Very Severe COPD, ASCENT COPD; major adverse CV events, MACE; myocardial infarction, MI; twice-daily, BID; long-acting beta2-agonist, LABA; serious adverse event, SAE; forced expiratory volume in 1 second, FEV1; inhaled corticosteroid, ICS; Food and Drug Administration, FDA; adverse event, AE; electrocardiogram, ECG; end of treatment, EOT; telephone contact, TC; COPD Assessment Test, CAT; hazard ratio, HR; patient-years, PY
Citation: Wise RA, Chapman KR, Scirica BM, et al. Long-term evaluation of the effects of aclidinium bromide on major adverse cardiovascular events and COPD exacerbations in patients with moderate to very severe COPD: Rationale and design of the ascent COPD study. Chronic Obstr Pulm Dis. 2018; 5(1): 5-15. doi: http://doi.org/10.15326/jcopdf.5.1.2017.0149
Introduction
It is estimated that up to 70% of patients with chronic obstructive pulmonary disease (COPD) suffer from cardiovascular (CV) comorbidities1 and approximately 30% of patients with COPD will die as a result of CV-related disease.2,3 Poor lung function and exacerbations have been associated with an increased risk of CV and cerebrovascular conditions, including myocardial infarction (MI), stroke, and sudden cardiac death.2,4,5 Patients with frequent exacerbations typically have a faster rate of decline in lung function, reduced daily activity, impaired health-related quality of life, increased airway inflammation, and higher mortality.6-11
Meta-analyses and observational studies have raised the question as to whether long-acting muscarinic antagonists (LAMAs) increase the risk of CV events.12,13 In contrast, large clinical trials have found no increased CV risk associated with LAMAs,14 including one study that included a subgroup of patients who experienced significant CV disorders during the trial (MI, cardiac arrhythmia or heart failure) for which they would have been excluded at baseline but who nonetheless remained in the trial.15 Despite the prevalence of CV-related risk factors in patients with COPD,1,16 patients with significant CV disorders are often excluded from COPD clinical trials.17 Thus, an evaluation of the safety of COPD treatments in this patient population is of particular clinical importance.
Aclidinium bromide is an inhaled LAMA approved for the long-term maintenance treatment of COPD-associated bronchospasm, including chronic bronchitis and emphysema.18 Short‑term studies (3 to 6 months in duration), including only patients with a low risk of CV events, found no increased risk of CV events19;however, longer-term studies, including patients at risk of CV events who are more representative of the COPD population in clinical practice, are necessary to evaluate CV safety effectively in patients with moderate to severe COPD. In addition, short-term studies with aclidinium have demonstrated decreased exacerbation rates after 3-6 months of treatment compared with placebo,20-22 suggesting that longer-term aclidinium treatment may further reduce exacerbations and associated hospitalizations.
The primary objectives of the long-term Aclidinium Bromide on Long‑Term Cardiovascular Safety and COPD Exacerbations in PatieNTs with Moderate to Very Severe COPD (ASCENT COPD) clinical outcome study are to evaluate the long-term effects of twice‑daily aclidinium 400 µg on major adverse CV events (MACE), overall safety, and COPD exacerbations in patients with moderate to very severe COPD who have a history or significant risk factors for CV comorbidities. The purpose of this manuscript is to describe the rationale and design of the ASCENT COPD study.
Methods
Study Design
ASCENT COPD is a Phase 4, multicenter, double-blind, randomized, placebo-controlled, parallel-group, clinical outcome study (NCT01966107) being conducted at 500 study centers in the United States and Canada. A full list of sites can be obtained from https://clinicaltrials.gov/. Approximately 4000 patients will be enrolled and treated until 122 adjudicated MACE occur, at which point the study will terminate. The study comprises a 2-week washout period, followed by a double-blind treatment phase in which patients are randomized 1:1 to aclidinium bromide 400μg or placebo twice daily, administered via a multidose dry powder inhaler (GenuairTM/Pressair®a)for up to 36 months (Figure 1). Patients who prematurely discontinue the double-blind treatment period (including those who experience a MACE) will participate in a post-treatment follow-up period for the remainder of the study duration. The follow-up period includes on-site and telephone visits for the monitoring of MACE, COPD exacerbations, concomitant medication use, and serious adverse events (SAEs).
The study is being conducted in accordance with the International Conference on Harmonisation/Good Clinical Practice guidelines and the Declaration of Helsinki. The protocol was approved by the institutional review board at each study center and all patients provide written informed consent before participating in any study procedures.
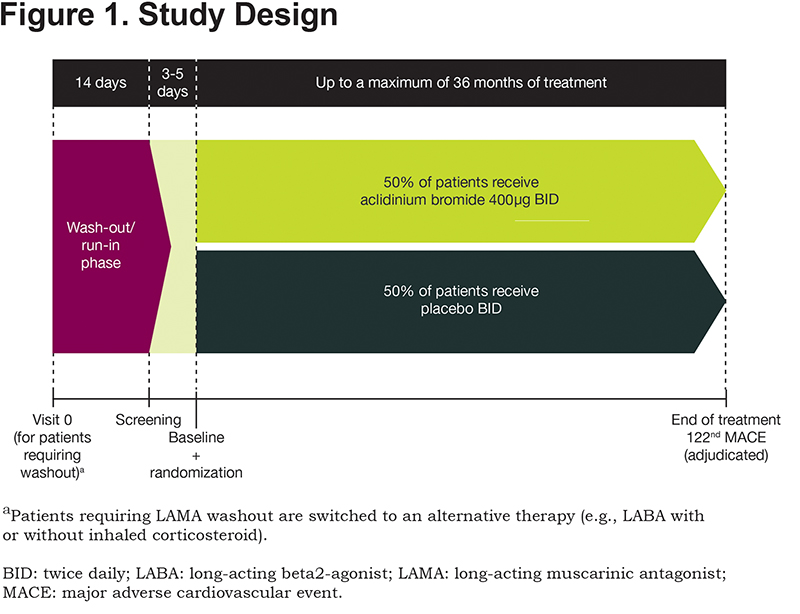
Patients
Male or female patients ≥40 years of age are eligible for inclusion if they have a diagnosis of stable, moderate to very severe COPD (as defined in the Global initiative for chronic Obstructive Lung Disease guidelines23), a post-bronchodilator forced expiratory volume in 1 second (FEV1) <80% predicted, FEV1/forced vital capacity ratio <70%, and are a current or former cigarette smoker with a smoking history of ≥10 pack years. Other key inclusion criteria include a history of significant CV or cerebrovascular disease and/or significant risk factors, defined by one or more of the following criteria:
1. Documented cerebrovascular disease (e.g., stroke or transient ischemic attack, carotid stenosis)
2. Documented coronary artery disease (e.g., angioplasty/stent/bypass, angina, MI)
3. Documented peripheral vascular disease or history of claudication
4. Two or more of the following CV risk factors, as determined by the investigator: male ≥65 years or female ≥70 years, diabetes, dyslipidemia, hypertension, waist circumference ≥40 inches for males and ≥38 inches for females, evidence of renal dysfunction (estimated Glomerular Filtration Rate <60), and microalbuminuria (defined as ≥30-300 µg/mg creatinine on a spot urine test or ≥30 mg creatinine on a 24-h urine test; renal dysfunction criteria were added after the initiation of patient enrollment).
When patient enrollment was initiated, patients were also required to have had at least 1 documented COPD exacerbation classified as moderate (requiring antibiotics, systemic corticosteroids) or severe (requiring hospitalization) in the 12 months prior to screening; however, this requirement was later removed from the inclusion criteria following a low rate of patient recruitment. Furthermore, the post-bronchodilator FEV1 inclusion criterion was amended from <70% predicted to <80% predicted.
Exclusion criteria include: treatment with triple therapy (LAMA, long-acting beta2-agonist [LABA], and inhaled corticosteroid [ICS]), respiratory infection or COPD exacerbation at screening or within 4 weeks prior to screening (patients can be rescreened after recovery), unstable or life-threatening COPD or CV disease (as determined by the investigator), comorbid lung disease other than COPD (e.g., asthma, cystic fibrosis, bronchiectasis, interstitial lung disease, pulmonary thromboembolic disease), planned lung transplant or lung volume reduction surgery, or malignancy requiring intervention (e.g., resection, radiation therapy, chemotherapy) within 5 years prior to screening.
Concomitant use of other COPD medications is permitted (including a LABA, with or without an ICS), except for anticholinergics and chronic treatment with regularly scheduled short-acting beta2-agonists, such as albuterol or salbutamol. Episodic use of sponsor-provided albuterol/salbutamol is allowed, but should not be taken within 6 hours prior to any visit. Oral or parenteral corticosteroids are permitted provided the patient is stable for at least 2 weeks before screening; however, they may be initiated, or the dose changed during the study, only at the discretion of the investigator.
Outcomes and Assessments
Safety
The incidence of MACE is specifically monitored during the study since both beta2-agonists and muscarinic antagonists have previously been associated with CV effects.24,25 The primary safety endpoint is time to first MACE (CV death, non-fatal MI, or non-fatal stroke) and the secondary safety endpoint is time to first MACE or other serious CV event of interest. All potential MACE, regardless of the gravity of the event, and all serious CV and cerebrovascular events of interest (see online data supplement) are assessed by a committee of independent physicians consisting of 2 cardiologists and 1 pulmonologist blinded to treatment assignment. The Clinical Endpoints Committee is responsible for adjudicating the following potential events as per the Food and Drug Administration (FDA) guidance paper26: 1) CV deaths; 2) Non-fatal MI; 3) Non-fatal stroke.
Patients who experience a MACE are immediately discontinued from treatment and will enter the post-treatment follow-up period. Patients who discontinue treatment early are contacted every 3 months for the first 2 years and every 6 months from the third year until the end of the study to obtain vital status information (MACE events, COPD exacerbations, SAEs, and concomitant medications). Adverse events (AEs) are recorded throughout the study, clinical laboratory tests are performed annually, and vital signs, physical examinations, and electrocardiograms are conducted throughout the study (Table 1). An independent Data Safety Monitoring Board periodically monitors and reviews relevant clinical safety data, including adjudicated MACE.
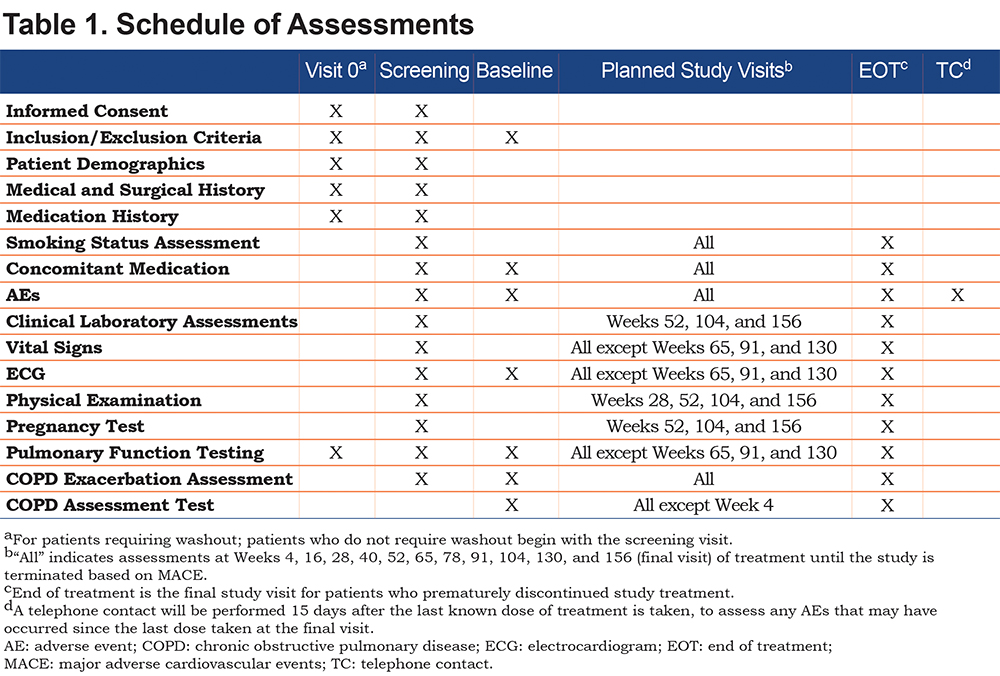
Efficacy
Based on the planned recruitment rate, by the time a total of 122 patients experience an adjudicated MACE, it is expected that almost all non-discontinued patients will have completed 1 year of treatment; however, the study does not require patients to be treated for 1 year to complete the study. The primary efficacy endpoint is the rate of moderate to severe COPD exacerbations per patient per year during the first year of treatment. A moderate exacerbation is defined as an increase in COPD symptoms (e.g., dyspnea, cough, sputum volume, sputum purulence) for at least 2 consecutive days that requires treatment with antibiotics and/or systemic corticosteroids. A severe exacerbation is defined as an increase in COPD symptoms for at least 2 consecutive days that leads to hospitalization (overnight stay at the hospital or emergency department) or death. The interval between 2 consecutive COPD exacerbations must be ≥7 days; if 2 COPD exacerbations occur within 7 days of each other, it will be considered as 1 event. Patients who experience a severe or recurrent COPD exacerbation may be treated with the addition of an allowed concomitant medication (ICS, oral or parenteral corticosteroid, LABA/ICS or roflumilast). However, if treatment with a LAMA is required, those patients will be discontinued from the study drug and entered into the post-treatment follow-up period.
The secondary efficacy endpoint is the rate of hospitalizations due to COPD exacerbations per patient per year during the first year of treatment. Additional efficacy evaluations include the change from baseline in morning pre-dose (trough) FEV1 and the COPD Assessment Test (CAT).27
Blinding
Randomization codes are generated by the sponsor and implemented using an interactive web response system. The system then assigns each patient with a sequential patient identification number that is used to identify the patient throughout the study and to obtain the kit to be dispensed to each patient at each visit.
Sample Size and Statistical Analyses
Sample Size
For the primary safety endpoint (time to first MACE), an estimated 122 patients with an adjudicated MACE are needed to achieve 90% power at the 5% significance level (2-sided) to rule out a hazard ratio (HR) of 1.8 in time to first MACE for aclidinium versus placebo. This assumes that the HR is 1.0 under the alternative hypothesis. The study was designed to exclude a pre-specified risk margin of 1.8 for the HR of MACE, which is in line with other CV outcome assessments in fields such as diabetes mellitus.28-30
The rate of MACE is assumed to be 1.9 MACE/100 patient-years (PY). Therefore, to generate the 122 MACE required, 6421 PY would be needed. Assuming a 40% premature treatment discontinuation rate and an average treatment duration of 1 year among those who discontinued and 2 years among those who did not, it was originally estimated that 4000 patients would be needed to obtain 6421 PY of follow-up.
The planned on-treatment analysis (including only events that occur while patients are exposed to study treatment) was later modified to an on-study analysis (including all events that occur while patients are in the study, irrespective of treatment exposure) with FDA agreement after initiation of patient enrollment. For the primary efficacy endpoint (rate of moderate to severe COPD exacerbations per patient per year), the sample size of 4000 patients after 1 year of treatment will provide ~89% power to detect a 14% reduction in the rate of moderate or severe COPD exacerbations per patient per year during the first year of treatment assuming a placebo rate of 0.8 exacerbations per patient per year and an overdispersion factor k of 0.67.31 The full analysis set will include all patients randomized to treatment and receiving at least 1 dose of study drug.
Statistical Analyses
The primary and secondary safety endpoints will be analyzed using a Cox proportional hazards regression model with baseline CV severity, smoking status, and treatment group as covariates; endpoints will be analyzed at the 95% confidence level based on an on-study analysis. An on-study sensitivity analysis will be carried out to assess the effect of missing follow-up time in patients who prematurely discontinued from the study. The primary and secondary efficacy endpoints will be analyzed based on a negative binomial regression model with treatment group, baseline ICS use, baseline COPD severity, history of at least 1 exacerbation in the past year, and smoking status as factors. If this model does not converge, a Poisson regression model with robust variance estimate using the sandwich method will be applied. Testing will be performed at the 0.05 significance level for all endpoints. The primary analysis on the primary efficacy endpoint will be based on on-treatment analysis, i.e., for those patients who prematurely discontinue treatment during the first year of the double‑blind treatment period and have follow-up assessments, the primary analysis described above will be based on the data collected before treatment discontinuation. Changes from baseline in morning pre-dose (trough) FEV1 and CAT will be analyzed using a mixed model for repeated measures adjusting for the corresponding baseline value (and pre- and post-bronchodilator FEV1 at screening for lung function), with baseline ICS use, smoking status, visit, and treatment group-by-visit interaction as fixed effect factors. Changes from baseline in the CAT score will also be evaluated in a responder analysis (≥2 units improvement)32 based on a logistic random effect model with baseline CAT total score as a covariate, and treatment group, baseline ICS use, smoking status, visit, and treatment group‑by-visit interaction as fixed effects and a random intercept to account for the variability between participants.
Discussion
The relationship between COPD and CV comorbidities is one of great clinical importance as CV events are a major cause of mortality among patients with COPD and much of the COPD population have comorbid CV conditions.1,5,16 Furthermore, as COPD exacerbations have been associated with CV events (see introduction), it is important to assess the long-term CV and cerebrovascular safety of any bronchodilator in patients with COPD. Despite this, patients with CV conditions are often excluded from COPD clinical trials.17 A number of studies have assessed CV safety of bronchodilators and have included patients with some CV risks (including obesity and hypertension); however, they have produced conflicting results.13-15,24,33-35 A pooled analysis of 2781 patients receiving aclidinium bromide for up to 6 months found no increased CV or cerebrovascular risk compared with placebo in patients with moderate to severe COPD.19 Of note, all of these studies have excluded patients who have had recent significant CV or cerebrovascular events. By including such patients, the ASCENT COPD study aims to specifically assess CV and cerebrovascular safety in a patient population with a higher CV and cerebrovascular risk profile.
Due to the low numbers of MACE in previous, shorter-term aclidinium studies, the impact of aclidinium on CV safety and exacerbations has not been fully established. Exacerbations are not only a debilitating occurrence for COPD patients, they are also associated with an increased risk for CV events.2,4 Thus, the evaluation of exacerbations as a primary outcome over 12 months provides an opportunity to better understand the cause, prevention, and treatment of exacerbations as well as how they relate to CV outcomes. Furthermore, with the exception of anticholinergics and the short-acting beta2-agonists albuterol/salbutamol, other COPD treatments are permitted in this study with consideration to preventing a high drop-out rate from patients in the placebo arm. This contrasts with most previous studies where no other long-acting bronchodilators were permitted. In addition, although patients with COPD often have CV comorbidities and risk factors, these were specified in the inclusion criteria for the ASCENT COPD study. The rationale for this was to include participants who would be most at risk for demonstrating a CV safety signal. Moreover, we anticipate that this population will have enough background CV events to allow a valid comparison of aclidinium and placebo.
The ASCENT COPD study has been designed to comprehensively evaluate CV safety and exacerbation rates in patients with moderate to very severe COPD treated with aclidinium compared with placebo. As this is an event-driven study design, the study will be terminated after a specific number of MACE have occurred (122 patients with an adjudicated MACE) rather than enrolling a specific number of patients and specifying an absolute study duration.
Although spirometry outcomes are widely used to assess the level of treatment benefits, they are not necessarily reflective of what physicians and patients view as beneficial.36 Evaluation of health status can facilitate a better understanding of how COPD symptoms are affecting a patient’s daily life, to better manage the disease. Although studies with aclidinium have demonstrated clinically meaningful and statistically significant improvements in health status and symptoms,21,37 the CAT results from the current study will indicate whether these improvements can be maintained over the course of several years alongside the eventual decline in lung function that occurs during the natural disease course of COPD.
Limitations of long-term placebo-controlled clinical studies of patients with COPD include an increased likelihood of discontinuations in patients receiving placebo that could lead to an underestimation of treatment effect. However, patients taking placebo were permitted to take LABAs with or without ICS and other non-anticholinergic medications to address this concern. This limitation was further addressed by the planned on-treatment analysis being modified to an on-study analysis.
Another limitation of this study is that although patients with a history of significant CV or cerebrovascular disease and/or significant risk factors were included in this study, patients with recent MI or stroke, or those with unstable angina, arrhythmia, or heart failure requiring hospitalization were excluded. Consequently, the results of the ASCENT COPD study cannot be generalized to those patients.
Current Status
The initial focus of the ASCENT COPD study was to investigate both efficacy and safety outcomes; however, the preliminary rate of patient enrollment (1605 patients in 22 months) was found to be 30% lower than expected. This lower than anticipated patient enrollment resulted from patients who had an exacerbation within the previous year prior to screening but were ineligible for study enrollment because they were also receiving triple therapy (ICS/LAMA/LABA). Considering the lower than expected enrollment, the inclusion criteria were revised to focus primarily on the safety outcomes, whilst retaining sufficient statistical power to permit analysis of the long-term effect of aclidinium on COPD exacerbations. This was achieved by amending the inclusion criteria to remove the need for patients to have had at least 1 documented moderate or severe COPD exacerbation. In addition, the range of post-bronchodilator FEV1 was increased from <70% to <80%. Following these changes, the rate of patient enrollment increased substantially.
The figure of 4000 randomized patients being required to achieve 122 adjudicated MACE was based on a MACE rate of 1.9 per 100 PY and a treatment discontinuation rate of 40%; however, the primary analysis was changed from on-treatment to on-study and the corresponding treatment discontinuation rate was lower than expected. Therefore, recruitment was terminated earlier because approximately 3500 randomized patients were estimated to provide the same on-study exposure as 4000 patients using the originally assumed treatment discontinuation rate. Recruitment was finalized in August 2016 and 3635 patients were randomized.
In summary, attaining the goals of the ASCENT COPD study poses several challenges and provides relatively unique study design elements to address them. The study was designed to evaluate both the safety (non-inferiority versus placebo for time to first MACE) as well as the efficacy (rate of moderate to severe COPD exacerbations) of aclidinium bromide in the same population. Recruitment challenges were overcome to preserve the primary safety objective by including patients with moderate to very severe COPD, regardless of the history of exacerbations; the size of the study population was sufficient to maintain the statistical power for efficacy endpoint evaluation. To ensure a definitive analysis of the safety outcome, this study was an event-driven study with a common close-out date rather than a fixed time exposure. The real-world external validity of the study was further strengthened by allowing patients to receive concomitant treatment with other non‑anticholinergic COPD drugs. Consequently, the ASCENT COPD study design can serve as a model for future studies of COPD treatments that aim to concurrently assess interrelated safety and efficacy outcomes reflecting COPD patient populations in clinical practice.
Conclusion
In conclusion, this long-term study specifically includes patients with CV and cerebrovascular comorbidities and will provide an understanding of the effects of aclidinium bromide on MACE and CV safety. In addition, the study is expected to provide further confirmation of the positive long-term effects of aclidinium bromide in reducing exacerbations whilst improving lung function and quality of life in patients with moderate to very severe COPD.
aRegistered trademarks of the AstraZeneca group of companies, for use within the United States as Pressair® and as GenuairTM within all other licensed territories.
Acknowledgments
The authors thank Richard Knight, PhD, of Complete Medical Communications, Macclesfield, United Kingdom, for providing medical writing support, which was funded by AstraZeneca in accordance with Good Publication Practice (GPP3) guidelines (Ann Intern Med. 2015;163:461-464). The authors also wish to acknowledge the assistance of Joaquim Delgadillo (AstraZeneca, Barcelona, Spain), Åsa Hellqvist, and Magnus Mattson (both AstraZeneca, Gothenburg, Sweden).
Declaration of Interest
RW reports grants and personal fees from Forest Laboratories and AstraZeneca during the conduct of the study and reports grants from Boehringer Ingelheim, GlaxoSmithKline, and Pearl Therapeutics, with personal fees from Boehringer Ingelheim, ContraFect, GlaxoSmithKline, Pulmonx, Roche, Spiration, Sunovion, Teva, Vertex, and Merck, outside the submitted work. KC has received compensation for consulting with AstraZeneca, Boehringer Ingelheim, CSL Behring, Genentech, GlaxoSmithKline, Grifols, Kamada, Merck Frosst, Novartis, Roche, and Sanofi and has undertaken research funded by Amgen, AstraZeneca, Baxter, Boehringer Ingelheim, CSL Behring, GlaxoSmithKline, Kamada, Novartis, Roche, and Grifols. KC has participated in continuing medical education activities sponsored in whole or in part by AstraZeneca, Boehringer Ingelheim, GlaxoSmithKline, Grifols, Merck Frosst, and Novartis. He is participating in research funded by the Canadian Institutes of Health Research operating grant, entitled: Canadian Cohort Obstructive Lung Disease (CanCOLD), and holds the GlaxoSmithKline-Canadian Institute of Health Research Chair in Respiratory Health Care Delivery at the University Health Network, Toronto, Canada. BMS reports grants from Eisai and Poxel, and personal fees from AstraZeneca, Lexicon, Boehringer Ingelheim, Biogen Idec, Forest Laboratories, Dr. Reddy's Laboratory, GlaxoSmithKline, Merck, Health@Scale, St. Jude's Medical, Elsevier, NovoNordisk, Allergan, Sanofi. DS has been a paid consultant to AstraZeneca. DLB has been on an Advisory Board for Cardax, Elsevier Practice Update Cardiology, Medscape Cardiology, Regado Biosciences and the Board of Directors for Boston Veterans Administration Research Institute and the Society of Cardiovascular Patient Care. He has chaired the American Heart Association Quality Oversight Committee and the Data Monitoring Committees of Duke Clinical Research Institute, Harvard Clinical Research Institute, the Mayo Clinic and the Population Health Research Institute. He has received honoraria from the American College of Cardiology (Senior Associate Editor, Clinical Trials and News, ACC.org), Belvoir Publications (Editor in Chief, Harvard Heart Letter), Duke Clinical Research Institute (clinical trial steering committees), Harvard Clinical Research Institute (clinical trial steering committee), HMP Communications (Editor in Chief, Journal of Invasive Cardiology), the Journal of the American College of Cardiology (Guest Editor; Associate Editor), the Population Health Research Institute (clinical trial steering committee), Slack Publications (Chief Medical Editor, Cardiology Today’s Intervention), the Society of Cardiovascular Patient Care (Secretary/Treasurer) and WebMD (CME steering committees). He has received research funding from Amarin, Amgen, AstraZeneca (including for his role as CEC Chair of ASCENT COPD), Bristol-Myers Squibb, Eisai, Ethicon, Forest Laboratories (for his role as CEC Chair of ASCENT COPD), Ischemix, Lilly, Medtronic, Pfizer, Roche, Sanofi Aventis and The Medicines Company and royalties from Elsevier (Editor, Cardiovascular Intervention: A Companion to Braunwald’s Heart Disease). He has been a site co-investigator for Biotronik, Boston Scientific and St. Jude’s Medical and has served as a trustee for the American College of Cardiology. He has conducted unfunded research for FlowCo, PLx Pharma and Takeda. EGG is an employee of AstraZeneca and a former employee of Almirall S.A., Barcelona, Spain. SD is an employee of AstraZeneca and a former employee of Forest Laboratories, LLC (a subsidiary of Allergan plc, formerly Actavis plc). CR and BS are employees of AstraZeneca.