Running Head: State of the Evidence for AAT Therapy in AATD
Funding support: Medical writing assistance was funded by CSL Behring
Date of acceptance: March 30, 2018
Abbreviations: alpha-1 antitrypsin deficiency, AATD; chronic obstructive pulmonary disease, COPD; alpha-1 antitrypsin, AAT; Food and Drug Administration, FDA; neutrophil elastase, NE; alpha-1 proteinase inhibitor, A1-PI; open label extension, OLE; forced expiratory volume in 1 second, FEV1; computed tomography, CT; quality of life, QoL; randomized controlled trial, RCT; St George’s Respiratory Questionnaire, SGRQ; total lung capacity, TLC; functional residual capacity, FRC; desmosine, DES; isodesmosine, IDES; graft versus host disease, GVHD
Citation: Brantly ML, Lascano JE, Shahmohammadi A. Intravenous alpha-1 antitrypsin therapy for alpha-1 antitrypsin deficiency: the current state of the evidence.
Chronic Obstr Pulm Dis. 2019; 6(1): 100-114. doi:
http://doi.org/10.15326/jcopdf.6.1.2017.0185
Introduction
Alpha-1 antitrypsin (AAT) is a polyfunctional protein of the SERPIN family.1 One of its best understood functions is the inhibition of neutrophil elastase (NE), whose principal function is the proteolytic degradation of matrix proteins.1,2 Genetic deficiency of AAT results in an imbalance in activity between the proteinase (NE) and the anti-proteinase (AAT).3 This imbalance can lead to the breakdown of elastin in tissues – principally in the parenchyma of the lung.3 As a result, individuals with severe alpha-1 antitrypsin deficiency (AATD), most commonly with the PI*ZZ, or PI*SZ genotypes or absence of the gene (PI*NullNull), are predisposed to early-onset emphysema.4 In addition, there is emerging evidence that AAT has several other functions that are thought to modulate inflammation in the lung and elsewhere.5
Despite the status of AATD as a rare disease, it is one of the most common fatal genetic disorders, and respiratory failure accounts for as much as 45% of deaths in never smoking AATD patients.6 AATD can also predispose to liver disease; as the protein is synthesized within hepatocytes, damage predominantly results from the aggregation of AAT caused by the PI*ZZ mutation and to a lesser extent by the PI*SZ variant.7 Liver disease is an often neglected aspect of AATD and it is interesting to note that, in a screening study, cirrhosis was found to account for 10% of deaths in AATD patients.8
Worldwide, there are estimated to be 181,894 and 1,269,054 individuals with the PI*ZZ and PI*SZ genotypes, respectively.9 However, only a small fraction of these individuals have been identified and most patients are identified long after symptoms begin.10 Importantly, non-affected and or mildly affected AAT-deficient individuals never develop life-threatening lung disease if early identification is coupled with smoking cessation and lifestyle counseling. Individuals with AAT deficiency and symptoms may benefit from AAT therapy.2 However, the initial diagnosis can be challenging as AATD-related chronic obstructive pulmonary disease (COPD) has no unique features that separate it from non-genetic COPD.11 To identify AAT deficient individuals, genetic testing is an absolute requirement. Furthermore, testing methods should take into consideration both quantitative levels of AAT and the patient’s genotype or AAT Pi-type to gain an accurate picture of the individual patient risk.12,13
The antitrypsin activity of AAT was first described in the 1950s by Jacobsson.14,15 The protein was later isolated by Laurell and Eriksson in the early 1960s, with some suggestion from case reports that low serum levels in patients were contributing to lung disease.16 Later, AAT was found to inhibit NE with a greater affinity, leading to the introduction of the nomenclature alpha-1 proteinase inhibitor (A1-PI). Since the discovery of a genetic deficiency in AAT, efforts have focused on isolating and purifying the protein from human plasma, in order to supplement patients with a deficiency.3 This has resulted in the development of products (Figure 1) aimed at raising serum levels of AAT in patients with AATD above a theoretical “protective” threshold of 11 µM. This level is equivalent to the 10th percentile of the AAT range of PI*SZ individuals, with epidemiological data suggesting a lower probability of COPD above this level.17,18 Proving the efficacy of exogenous administration of AAT in AATD beyond the biochemical elevation of anti-elastase capacity, i.e., the clinical efficacy to slow emphysema progression, has proved to be more difficult. This has been in part due to the orphan status of the disease and the previously discussed barriers to diagnosis, but has also been confounded by a lack of sensitive clinical study endpoints to monitor disease progression.19 As a result, the treatment of AATD with intravenous AAT therapy has remained a controversial topic and has hampered efforts to raise awareness of the disease.
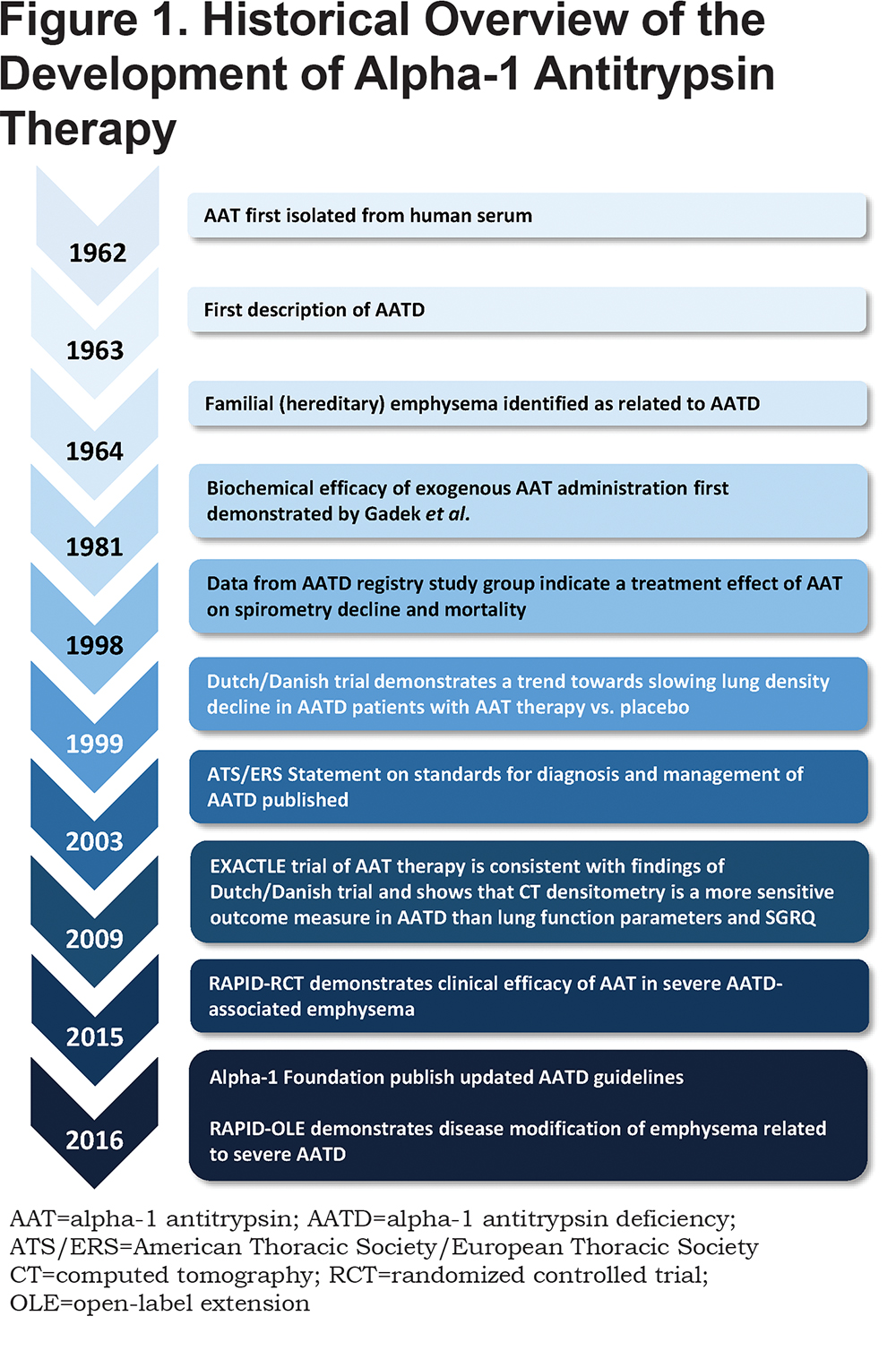
This review aims to update a previous publication by Tonelli and Brantly (2010)2 in light of the publication of data from several recent clinical studies, notably, data from the RAPID clinical trial program – the largest clinical trials of AAT therapy on emphysema progression in AATD completed to date. The present review will focus on and reassess the clinical evidence for the efficacy of exogenous AAT therapy administration to treat patients with emphysema related to severe AATD. The evidence for other clinical uses of AAT therapy will also be covered.
Search Strategy and Selection Criteria
For the present narrative review, literature was sourced from the references listed in the Tonelli and Brantly2 paper. PubMed searches were performed to identify clinical trials as well as clinical and pre-clinical studies of AAT therapy published since 2010 that were not included in the original paper. Search terms included: “alpha-1 proteinase/alpha-1 antitrypsin AND emphysema”; “AATD AND clinical trial”.
Current State of the Evidence for Alpha-1 Antitrypsin Therapy
Biochemical Efficacy
The biochemical efficacy of AAT therapy has been well established in numerous studies – doses of 60 mg/kg and above raise serum levels above the theoretical protective threshold of 11 µM and increase the inhibition of NE.3,20-28 To date, only the 60 mg/kg/wk dose has been proven to maintain AAT serum levels consistently above 11 µM. Following longer dosing intervals and higher doses (i.e., 120 mg/kg biweekly and 250 mg/kg monthly), serum AAT levels were found to fall below the therapeutic threshold before the next scheduled dose.21,22
Recent studies evaluating the biochemical efficacy of AAT since the publication of the previous review2 are shown in Table 1. A recent pharmacokinetic study by Campos et al (2013)26 comparing 120 mg/kg versus 60 mg/kg AAT weekly demonstrated that the higher dose achieved a significantly higher trough level (27.7 versus 17.3 µM). However, clearance of the higher dose was faster: half-lives were 184.3 hours versus 212.7 hours for 120 mg/kg and 60 mg/kg, respectively.26 The Campos et al study demonstrated no difference in adverse event rates between the 2 doses. This latter observation is supported by data from the RAPID clinical trial program; patients received a higher bi-weekly dose of 120 mg/kg to cover periods where they were unable to receive weekly infusions, e.g., for vacations.29 Both the 60 and 120 mg/kg doses achieved serum levels above 11 µM and were associated with similar adverse event rates to placebo, with no serious adverse events deemed to be treatment-related.29 Recently, a sub-analysis of the RAPID clinical trial program confirmed the stability of weight-based dosing of 60 mg/kg/wk in achieving the 11 µM threshold, as higher/lower weight than the average was predicted to have little effect on achieved serum levels.30
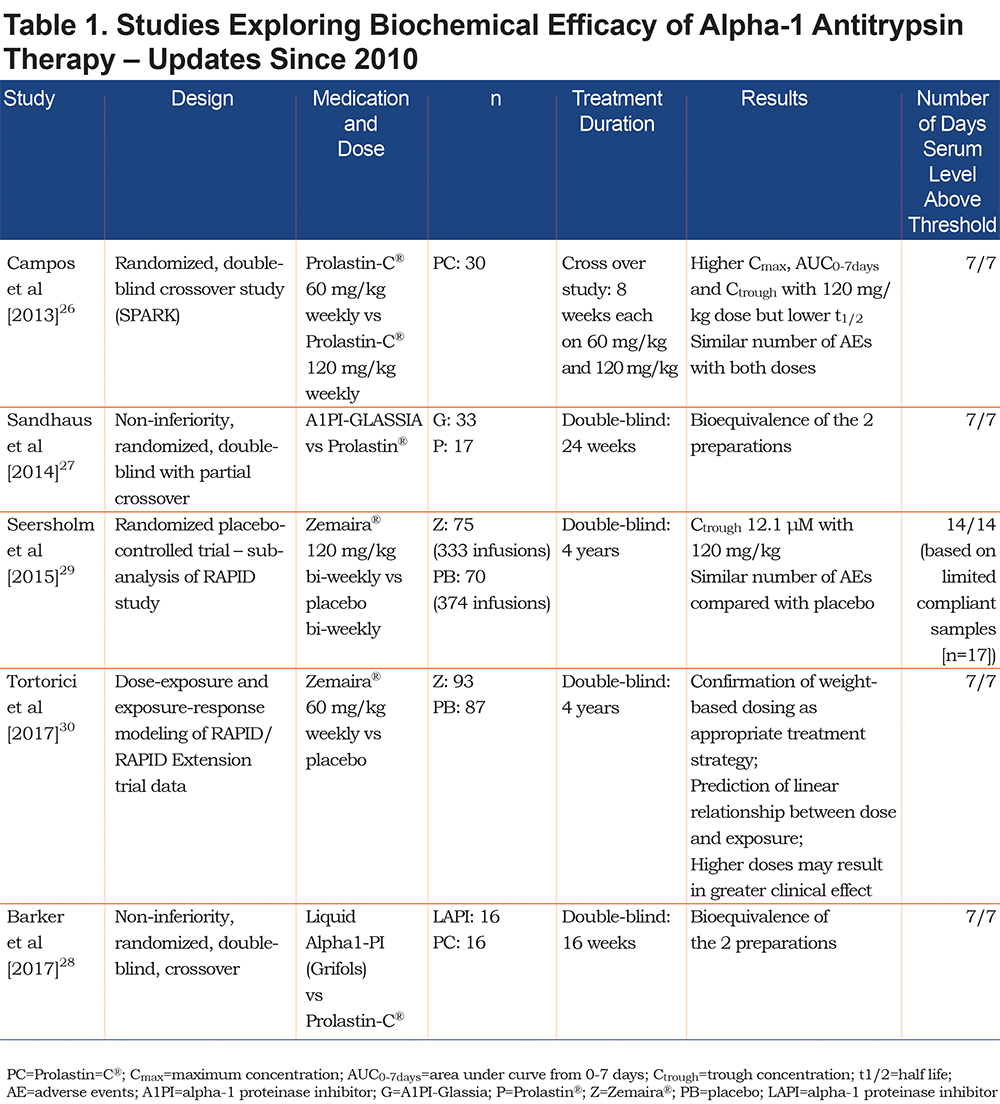
Despite most biochemical studies focusing on achieving the putative 11 µM protective threshold, there remains controversy surrounding the clinical meaning of this level – this topic is discussed further in the questions section. In addition, while higher doses of AAT therapy may be beneficial in some patients, the long-term safety of higher doses remains unanswered and ongoing studies will seek to address this issue.31
Clinical Efficacy in Lung Disease
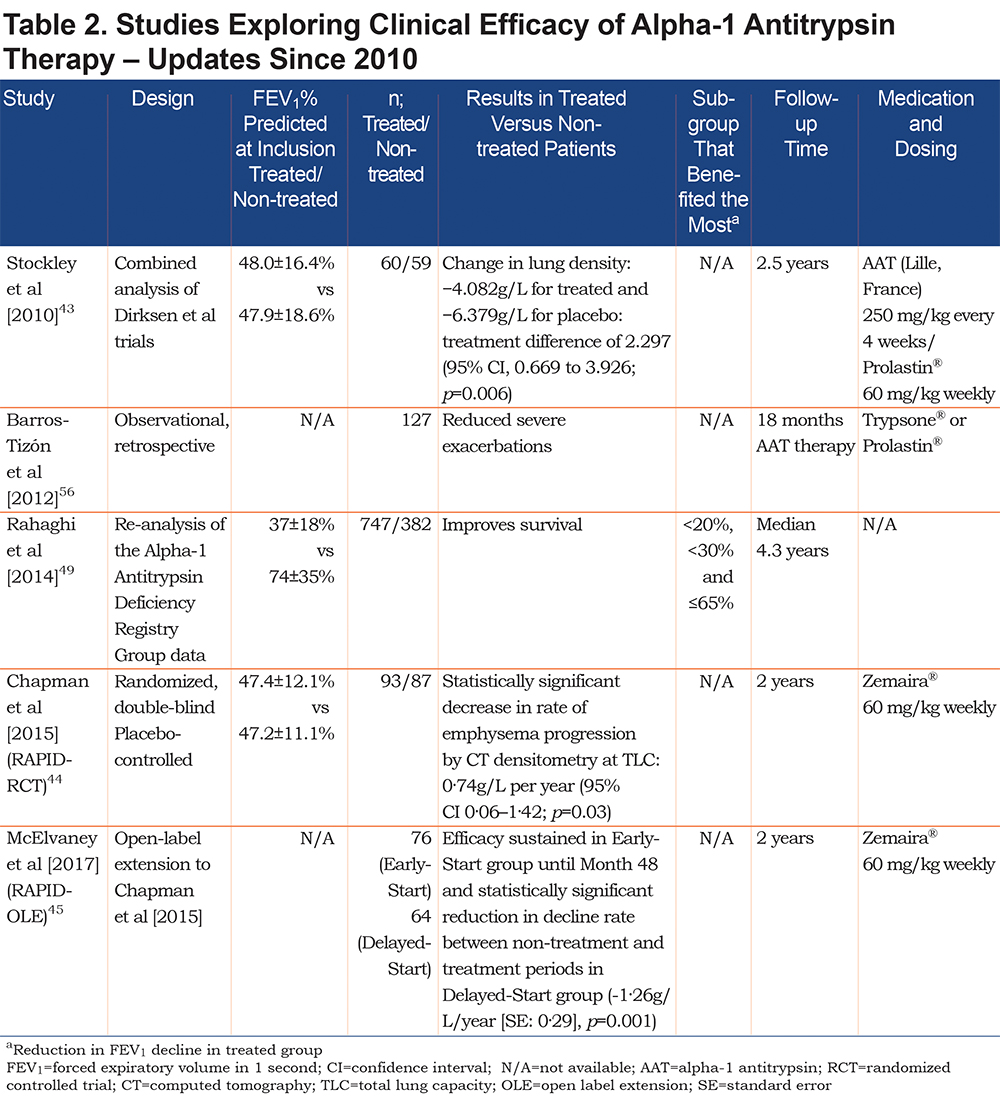
In attempting to demonstrate the clinical efficacy of AAT therapy, various clinical endpoints have been assessed, including spirometry (forced expiratory volume in 1 second [FEV1]), exacerbations, lung density measured by computed tomography (CT), quality of life (QoL) and mortality. The evidence for the efficacy of AAT therapy on these endpoints is discussed below. Recent studies published since 2010 that have investigated the clinical efficacy of AAT therapy are shown in Table 2.
Effect on Spirometry
To show a statistically significant effect of AAT therapy on emphysema progression using FEV1 in a randomized, placebo-controlled trial, it was originally estimated that around 300 patients would need to be followed over 3 years.19 This represented a significant logistical challenge, owing to the previously discussed difficulties surrounding diagnosis coupled with the orphan status of the disease. Observational and registry studies were, therefore, the best approach to gain an indication of the natural history of AATD and efficacy of treatment. Several studies noted a significant effect of AAT therapy on FEV1 decline, most notably within specific FEV1 ranges.32-36 The largest pool of spirometry data published in 1999 by the Alpha-1 Antitrypsin Deficiency Registry Study Group suggested that AAT therapy was effective between the FEV1 range of 35% and 49% predicted.33
Despite evidence of a therapeutic effect on FEV1 in observational or registry studies, clinical studies have failed to demonstrate a significant effect, in part due to smaller sample sizes and length of follow-up.37,38 A meta-analysis by Chapman et al (2009) pooled data from 5 studies (N=1509) and concluded that the rate of FEV1 decline was 23% less in patients receiving AAT therapy, with the difference predominantly seen in patients with FEV1 30%–65% predicted.39 No significant effect was shown with values above or below this range. However, as FEV1 has low sensitivity in AATD,38 these data do not rule out the possibility that AAT is effective in more or less advanced disease.
Wencker et al (2001) found that the rate of lung function decline may be more important than initial disease severity when attempting to demonstrate treatment efficacy of AAT therapy.34 Contrary to findings from other studies, as described above, statistical significance for AAT treatment versus non-treatment was only reached in patients with baseline FEV1 >65%. However, this was due to the majority of these patients exhibiting very fast FEV1 decline and consequently showing extremely large responses to treatment. This study also highlighted that individual decline in FEV1 can be non-linear. Furthermore, a recent analysis provides further evidence of the insensitivity of FEV1 to detect changes in emphysema progression and AAT treatment effect in AATD40; this will be discussed in more detail later in the review.
Overall, although an effect of AAT therapy on spirometric decline has been demonstrated in observational studies, use of this endpoint in clinical trials has been limited due to the need for larger numbers of patients and longer follow-up as indicated by power calculations.19,37 Therefore, endpoints with a higher sensitivity are required to assess the effect of AAT therapy on emphysema progression in AATD.
Effect on Lung Density
Although a treatment effect of AAT therapy was suggested by observational cohort studies, there are several limitations to these data, primarily the potential for investigator bias. High-quality evidence from a randomized controlled trial (RCT) was needed to confirm a treatment effect, and a key consideration for clinical studies was the use of highly sensitive and reliable endpoints.41 CT lung densitometry has been shown to correlate with both pathological changes in the lungs and mortality, and was therefore approved by the FDA for use as a surrogate endpoint for emphysema progression in AATD.42
The Dirksen et al study (1999) of 26 Danish and 30 Dutch smokers was the first clinical trial to investigate the utility of CT-measured lung density in AATD.37 Although the trial was not powered to detect a statistically significant effect of treatment, a trend in favor of AAT therapy was shown, with an annual loss of lung tissue, as measured by CT, of 2.6 ± 0.41 g/L/yr for placebo compared with 1.5 ± 0.41 g/L/yr for AAT (p=0.07). Power calculations indicated that a cohort of approximately 130 patients would have been sufficient to show a statistically significant effect. The subsequent EXACTLE study sought to further explore and refine the use of CT and compared the sensitivity of CT with other clinical outcome measures.38 Overall, CT was shown to have a higher sensitivity index score than lung function parameters, exacerbation frequency and QoL, as determined by the St George’s Respiratory Questionnaire (SGRQ). The results from EXACTLE mirrored that of the Danish/Dutch trial: all methods of CT analysis favored AAT therapy versus placebo with p-values ranging between 0.049 and 0.084.38 In 2010, a pooled analysis of the 2 RCTs found a statistically significant effect on lung density decline; overall decline rates were −1.73 and −2.74 g/L/yr, for AAT therapy and placebo, respectively (p=0.006)43
Recently, data from the RAPID clinical trial program, the first adequately powered clinical trial in AATD, were published.44,45 RAPID-RCT was a multi-center, double-blind, placebo controlled trial of AAT therapy that recruited 180 AATD patients from 28 centers worldwide. After 2 years, non-U.S. patients were eligible to participate in an open-label extension phase (RAPID-OLE). During the 2-year extension study, patients who were previously assigned placebo in RAPID-RCT were switched to active treatment. In total, 140 patients (intention-to-treat population: 76 Early-Start and 64 Delayed-Start) continued treatment with AAT therapy, with 121 completing the full 4 years.
Over 2 years of the RAPID-RCT study, the annual rate of lung density decline was –2.19 g/L for placebo versus −1.45 g/L at total lung capacity (TLC; p=0.03).44 While a significant reduction in lung density decline rate was observed at TLC, results at functional residual capacity (FRC) alone or at TLC + FRC did not reach statistical significance. However, this difference can be explained by higher measurement error in CT scans at lower lung volumes.46 In the RAPID-OLE analysis; between Day 1 and Month 24, the annual decline rate at TLC was 33% higher in Delayed-Start patients (Placebo – AAT) compared to Early-Start patients (AAT – AAT): −2.26 g/L/year versus −1.51 g/L/year; p=0.021. Treatment efficacy of AAT therapy was also maintained over 4 years in the Early-Start group. When patients in the Delayed-Start group were switched from placebo in RAPID-RCT to active treatment in RAPID-OLE, a statistically significant decrease or inflection point in the rate of lung density decline was observed.45 However, these patients failed to catch-up with the Early-Start group, due to the permanent loss of lung tissue,45 consistent with a “disease-modifying” effect of AAT therapy in AATD, i.e., a change in the natural history of disease progression.41
Recent CT efficacy data also challenge findings from observational studies showing an effect of AAT therapy on spirometric (FEV1) decline at specific levels of baseline disease severity. A regression analysis comparing AAT therapy and placebo in all patients in RAPID-RCT found the treatment advantage with AAT (reduction in lung density decline rate) to be flat across all levels of baseline FEV1 impairment (between 27% and 79% predicted).40 These data suggest that efficacy of AAT therapy is independent of baseline FEV1 and disease severity, although further data are needed to support these findings.
Mortality
Emphysema related to AATD is a slowly progressing disease that is often fatal. To date, a mortality benefit of AAT therapy has not been confirmed in a clinical trial. Previous clinical trials have not been sufficiently powered to directly assess the effect of AAT therapy on mortality, with much larger sample sizes and longer duration placebo-controlled trials needed, particularly as a recent study by the U.K. ADAPT registry suggests that the majority of deaths occur after 4 to 9 years of follow-up.47 Nevertheless, observational studies have correlated lower lung density with reduced survival, providing an indirect indication of a mortality benefit with AAT therapy.47,48 The AATD registry suggested a beneficial effect on survival: mortality rate was statistically lower in the patients receiving AAT therapy compared with non-treated individuals, an effect predominantly observed in patients with an FEV1 <50% predicted.33 A recent re-analysis of these data have shown a survival improvement also in patients with low baseline FEV1, <20% and <30% predicted.49 However, a significant confounding factor for the AATD registry study was that the socioeconomic status of the treatment versus non-treatment cohorts could not be accounted for. In addition, a post-hoc extrapolation analysis of data from the RAPID clinical trial program suggests that AAT therapy can delay progression to terminal lung function (~20 g/L; defined as lung transplant, severe respiratory crisis or death) by 5.6 years.45
Effect on Biomarkers
There is increasing interest in the use of biomarkers to predict disease outcomes in COPD. A recent example is fibrinogen, increased levels of which have been shown to predict exacerbation risk and mortality.50
The main pathological process in AATD, the degradation of elastin, yields desmosine and isodesmosine (DES/IDES), biomarkers that have been investigated for their potential use in tracking disease progression and treatment efficacy in AATD.51 Previous studies have shown significantly lower levels of DES/IDES in patients receiving AAT therapy compared to patients not receiving treatment.52 Levels were also shown to be significantly higher in AATD patients compared to non-AATD controls, with those receiving AAT therapy statistically closer to normal individuals. The validity of DES/IDES as a biomarker in AATD is supported by a recent publication by Ma et al (2016) concerning the measured level of DES/IDES in patients during the RAPID clinical trial program.53 During RAPID-RCT, the levels of DES/IDES were statistically lower at all time points in the treated group compared with placebo. This effect was maintained in patients who originally received active treatment in RAPID-RCT (the Early-Start group) over the full 4 years of RAPID-RCT and RAPID-OLE. In the Delayed-Start group, switching to active treatment at month 24 was associated with a significant reduction in DES/IDES at months 36 and 48. A weak but significant correlation between changes in DES/IDES and the CT densitometry data at TLC was also shown (R=−0.256, P=0.005). DES/IDES show promise in providing a low-cost estimate of the severity of disease in AATD patients. However, such are the extremely small quantities of DES/IDES involved (in the nanogram per milliliter range) that highly sensitive analytical equipment is required. Further development of an assay for use in clinical trials is therefore necessary.
Other Endpoints
Other clinical endpoints have been investigated in clinical studies of AAT therapy, these include exacerbation frequency/severity and QoL, as assessed by the SGRQ.
Lung exacerbations are a significant health care burden in AATD and contribute substantially to poor outcomes and QoL.54 Two observational studies have reported beneficial effects of AAT therapy in reducing exacerbation frequency55,56; Barros-Tizon et al (2012) demonstrated a reduction in all (p<0.05) and severe (p<0.01) exacerbations in a cohort of 127 patients. This was reported to equate to a significant reduction in health expenditure per patient and was more apparent in patients who had previously experienced an exacerbation.56 However, although the cohort of patients were reportedly well-matched socioeconomically, the absence of a control group and the retrospective manner of the investigation are significant limitations. To date, a beneficial effect of AAT therapy on exacerbations has not been confirmed in several clinical trials.37,38,44 However, as AAT has significant anti-inflammatory properties,57 treatment may have a greater impact on exacerbation severity, rather than frequency. The EXACTLE clinical trial studied the effect of AAT therapy on exacerbations and reported an improvement in the qualitative severity of exacerbations following AAT treatment38; however, no specific details of this effect are provided. The effect of AAT therapy on exacerbations in AATD has yet to be confirmed and future clinical trials will have to be appropriately designed to fully investigate this.
The SGRQ is a commonly used QoL assessment tool in respiratory medicine. The SGRQ was included as a secondary endpoint in several clinical trials of AAT therapy completed to date. However, as no trials to date have been sufficiently powered to fully assess the effect of therapy on the SGRQ, owing to its low sensitivity index in AATD,38 they have failed to demonstrate a significant effect of therapy versus placebo.38,44,45
Other Uses for Alpha-1 Antitrypsin
Transplant
AAT therapy has been investigated for its potential in treating graft versus host disease (GVHD) in organ transplantation. Current evidence based on mouse models has shown that AAT treatment significantly reduces pro-inflammatory cytokines in GVHD post bone-marrow transplantation and can induce immune tolerance following pancreatic islet allograph transplantation.58,59 Due to the well-established safety profile of AAT therapy in humans, the treatment is currently being tested in Phase 1/2 open-label studies of AAT therapy in steroid refractory acute GVHD.60,61
Diabetes
Due to its anti-inflammatory activity and ability to protect pancreatic islets from immune destruction, AAT therapy has been investigated for its role in slowing the progression of recent-onset type-1 diabetes mellitus in children.62 Recent results from a Phase 1/2 clinical trial were promising with at least 8 out of 24 patients showing a discernable response in terms of glycemic control.62
Questions
Answers to the following questions are based on evidence collated within this review, with updates on the 2010 Tonelli and Brantly paper.
1. Why have clinical trials focused on CT lung density rather than mortality?
Despite limitations associated with the AATD registry study, its data are suggestive of a mortality effect – questions may therefore be asked regarding why this has not been the focus of clinical trials. As discussed earlier in the review, to demonstrate a discernable effect on mortality in a clinical trial, a large number of patients would potentially need to be followed for up to 10 years.47 There are several factors that limit the feasibility of this, including difficulties in recruiting sufficient numbers of AAT-naive patients, ethical implications associated with administering placebo for this length of time and the overall expense of such a study. Due to these limitations, clinical trials have utilized CT lung density – a more sensitive outcome measures that correlates with mortality.47,48 By utilizing CT densitometry, clinical trials have demonstrated the treatment efficacy of AAT therapy in AATD37,38,44,45
In addition, extrapolation analyses, such as the post-hoc analysis of the RAPID clinical trial program, suggest that AAT treatment may extend the time to a terminal respiratory event (e.g., lung transplantation or death). However, this evidence of a mortality benefit is indirect, and direct mortality data has been highlighted by some as key to supporting clinical efficacy.63 Therefore, further mortality data is needed and long-term terminal lung density data could support the proposed life-extending effect of AAT therapy.
2. Is there now sufficient evidence of clinical efficacy to recommend AAT therapy?
Half a century after AAT was first isolated, there is now evidence that administration of AAT therapy raises serum levels and increases anti-elastase capacity, and that this biochemical effect translates into a measurable decrease in lung-function decline. Furthermore, it has now been shown that AAT therapy modifies the course of disease by slowing the rate by which lung tissue is degraded. Based on what is now known about the natural history of AATD and how AAT therapy can potentially alter the course of the disease, there is a sound basis for symptomatic patients with a genetically confirmed deficiency to be prescribed AAT therapy as outlined in the Alpha-1 Foundation Guidelines64 (Table 3). However, it should be noted that, with a recent sub-analysis of RAPID-RCT, there is emerging evidence to suggest that AAT therapy is effective at all levels of FEV1 impairment, and that treatment is therefore beneficial in both early- and late-stage disease.40 However, further data are needed to support this. Nevertheless, as evidence from the RAPID trial program demonstrates that lung tissue lost during periods of inactive treatment cannot be regained, earlier intervention may be beneficial for many AATD patients. Although the exact timing of treatment initiation is a decision for the treating physician, based on the patient’s symptoms and the rate of deterioration in health status, the potential for earlier intervention is reflected in the Alpha-1 Foundation Guidelines. While a firm recommendation is provided for initiating AAT therapy in patients with FEV1 ≤65% predicted, it is also recommended that the possibility of commencing therapy be discussed at all levels of spirometric decline above this threshold.64

3. Should we expect a significant effect on quality-of-life outcomes?
The SGRQ was designed to assess the effect of interventions that improve symptoms in COPD; however, while there is evidence that AAT therapy can slow disease progression, it cannot regain the lung tissue already lost pre-treatment, as demonstrated by the RAPID clinical trial program data.44,45 Moreover, the question of whether AAT therapy has an effect on exacerbations, a significant contributor to poor QoL outcomes, has yet to be conclusively determined. As the treatments and outcomes differ between COPD overall and AATD-related emphysema, there is an argument to be made that a QoL measure specific to AATD is needed to fully assess this question.
4. Is 11 µM the right “protective threshold” below which patients should receive AAT therapy?
As discussed previously,2 this target includes the endogenous, functionally deficient protein (in for example PI*ZZ patients) and is a measure of the total antigenic, rather than the active level. Nevertheless, by utilizing the 11 µM threshold as a point of reference, both observational studies and clinical trials (as summarized within this review) have demonstrated that elevating antigenic AAT levels above this level with the 60 mg/kg/wk dose exerts a significant clinical effect. However, to date clinical studies have focused on confirming the efficacy of the standard 60 mg/kg/wk and dose-optimization has yet to be explored. The recent pharmacometric analysis of the RAPID trials by Tortorici et alprovides the first tentative indication that higher doses may yield a greater clinical effect. In the RAPID trials, due to higher body weight and different volumes of distribution, some patients had higher exposure levels of AAT and these patients tended to have greater reductions in the rate of lung density decline.44 When the data were modeled, a linear association between exposure and response (reduction in decline rate) was predicted – suggesting that higher doses may be associated with a greater clinical response.30 The authors, Tortorici et al, acknowledge that it is unlikely for the relationship between exposure and response to be completely linear and that a plateau would be expected. However, the plasma level at which this plateau occurs is likely to be beyond the traditional 11µM threshold,30 which is the target for the 60 mg/kg/wk dose. The potential for greater efficacy on the rate of lung density decline with higher doses prompted the European Medicines Agency to request a post-marketing study following on from the RAPID trials to evaluate the efficacy of both 60 mg/kg/wk and 120 mg/kg/wk doses.65 In addition, another clinical trial of AAT therapy efficacy on lung density with the same high/low dose design was also planned and is now active.31 Moreover, a recently completed small-scale clinical trial provides evidence of increased biological activity with higher doses. The study found that normalizing AAT serum levels in severe AATD patients with doses of 120 mg/kg/wk resulted in a significant reduction in inflammatory markers versus the 60 mg/kg/wk dose.66 However, further research is required to establish whether increased anti-inflammatory activity translates into improved clinical outcomes. Therefore, as clinical evidence for increased efficacy with higher doses is scarce, doses >60 mg/kg/wk are not currently licensed and this is unlikely to change until clinical efficacy and long-term safety are demonstrated in a large-scale clinical trial.
5. What is the correct dosing interval?
As summarized within this review, several studies have experimented with extended and individualized dosing intervals. However, only the 60 mg/kg weekly dose has been shown to consistently maintain AAT serum levels above the 11 µM threshold, and serum AAT levels have been shown to decrease below 11 µM 1–2 days prior to the next dose with 120 mg/kg biweekly.21 Despite the previously discussed limitations with the 11 µM threshold, the clinical implications of not consistently raising levels above this threshold are uncertain, as most clinical efficacy data to date are based on the standard 60 mg/kg/wk dose. However, recent data from the RAPID clinical trial program demonstrate that longer dosing intervals may be beneficial in certain situations, e.g., to cover vacations, and importantly, that higher doses are not associated with increased adverse events.29 Nevertheless, due to a lack of clinical efficacy data based on alternative dosing strategies, the standard 60 mg/kg/wk dose should be the mainstay of treatment at the present time.
6. How should patients on AAT therapy be followed?
There is a lack of a clear consensus on this issue. As discussed previously,2 one firm recommendation from the original American Thoracic Society/European Respiratory Society statement is to measure FEV1 on a yearly basis.12 There has previously been limited evidence for tracking AATD disease progression with CT densitometry; however, the RAPID clinical trial program data provide a sound basis to recommend the use of CT densitometry. The recently published Alpha-1 Foundation AATD guidelines provide a firm recommendation that all newly diagnosed, symptomatic AATD patients should have a baseline chest CT scan.64 However, serial chest CT scanning is not recommended owing to radiation dose and uncertainty regarding how exactly the results should be interpreted clinically. This recommendation may change in years to come as lower-radiation scanning is introduced and the value of quantifying the level of disease progression is fully understood.
7. Should individuals with PI*MZ and PI*SZ receive AAT therapy?
There is limited rationale for treating patients with intermediate deficiency (i.e., PI*MZ individuals),67 and there is currently no evidence to support this use of AAT therapy – this is reflected by a firm stance in the latest guidelines (Table 3).64 However, if a patient is found to have the PI*MZ genotype, the chance of a direct family member showing homozygosity is significantly higher (owing to Mendelian inheritance).12,64 Therefore, familial testing of PI*MZ individuals is advised. The PI*SZ genotype is considered to be a severe deficiency variant64; however, clinical presentation is highly variable and PI*SZ individuals make up a small percentage (as little as 4%) of the lung pathologies associated with AATD.68 The decision of whether to treat PI*SZ patients therefore remains at the discretion of the treating physician based on AAT serum levels and clinical presentation. Furthermore, in recent years indications for treatment have moved away from narrow ranges of genotypes due to the high heterogeneity of the SERPINA1 gene, demonstrated by the large numbers of novel pathogenic mutations that continue to be discovered. Physicians are therefore encouraged to take a holistic approach and look at all aspects of AATD, from AAT serum levels to lung-function parameters and smoking status.
Areas for Future Research
1. Identifying an average terminal lung density threshold in AATD and emphysema to help quantify treatment-advantage with AAT therapy.
2. Dose optimization: now that the clinical efficacy of AAT therapy in AATD has been demonstrated, efforts should focus on identifying the optimal dosage/dosage interval.
3. Further study is required to confirm efficacy of AAT therapy in both early- and late-stage disease suggested by the RAPID-RCT data.
Conclusions
It is pleasing to see significant steps forward in the understanding of AATD. In the 8 years since the publication of the previous review, the RAPID clinical trial program has demonstrated that AAT therapy can significantly reduce lung density decline and is disease modifying. Nevertheless, despite the significant progress there remains doubt among some parties regarding the meaning of CT data and there is work required to conclusively link the reduction in rate of lung density decline to a mortality benefit. While it is difficult to demonstrate a mortality benefit of AAT therapy in a clinical study, data from the RAPID trial program indicate that treatment with AAT may delay the time to terminal lung function. Furthermore, the use of AAT therapy continues to be researched in other therapy areas, particularly in transplant and type-1 diabetes, in part due to its highly positive safety profile. Results from ongoing clinical trials in these areas are eagerly awaited.
Declaration of Interest
The authors have no conflicts of interest to declare. Medical writing assistance was provided by Steven Foster of Meridian HealthComms Ltd (Plumley, United Kingdom), funded by CSL Behring.