Running Head: Bronchiectasis in AATD, CVI and PCD
Funding Support: Funding provided by the COPD Foundation.
Date of acceptance: December 28, 2018
Abbreviations: alpha-1 antitrypsin deficiency, AATD; common variable immunodeficiency, CVI; primary ciliary dyskinesia, PCD; standard deviation, SD; forced expiratory volume in 1 second, FEV1; forced vital capacity, FVC; Bronchiectasis Research Registry, BRR; nontuberculous mycobacteria, NTM; intravenous immunoglobulin, IVIG
Citation: Eden E, Choate R, Barker A, et al. The clinical features of bronchiectasis associated with alpha-1 antitrypsin deficiency, common variable immunodeficiency and primary ciliary dyskinesia--results from the U.S. Bronchiectasis Research Registry. Chronic Obstr Pulm Dis. 2019; 6(2): 145-153. doi: http://doi.org/10.15326/jcopdf.6.2.2018.0156
Online Supplemental Material: Read Online Supplemental Material (94KB)
Introduction
Note: The abstract from this manuscript was presented at the American Thoracic Society International Conference in May 2018 and at the World Bronchiectasis Conference in July 2018.
The Bronchiectasis Research Registry (BRR) provides a database for studying associations between the reported characteristics of non-cystic fibrosis bronchiectasis in a U.S. population.1 Bronchiectasis may be associated with severe alpha-1 antitrypsin deficiency (AATD)2 —a condition with defective protease inhibition, common variable immunodeficiency (CVI)3 —a mixed group of conditions with a defect in humoral immunity and primary ciliary dyskinesia (PCD)4 with a defect in the airway clearance of secretions. The purpose of this report is to compare and contrast the clinical characteristics of bronchiectasis associated with these rare conditions with idiopathic bronchiectasis. By comparing and contrasting these conditions against a control subgroup, the aim of this report is to highlight these differences and to generate hypotheses about the pathogenesis of bronchiectasis in the different conditions.
Methods
This study used baseline data that were extracted from the BRR, a centralized database of individuals with bronchiectasis identified at 13 clinical sites throughout the United States.1 (see online supplement) The institutional review board of each participating center approved the study. Participants were enrolled from 2008 and follow-up data may be submitted yearly for as long as participants consent to remain in the Registry. At the time of this report, January 2017, 2170 participants had been enrolled. Adults 18 years or older with a physician-established diagnosis of non-cystic fibrosis bronchiectasis were eligible for inclusion. After providing informed consent, enrolled participants and/or their medical records were queried by a study coordinator or principal investigator to obtain demographic, clinical, laboratory, microbiologic, spirometric, radiographic, and treatment information using standardized recording forms. Cystic fibrosis status was established based on clinical history, sweat chloride and/or genetic testing results at the time of enrollment into the BRR. Those with known cystic fibrosis were ineligible for participation. Exacerbations were recorded as historical information and based on the answer to the BRR baseline question: “Has the patient experienced an exacerbation of bronchiectasis within the past 2 years?” Investigators at each center had available the definition of an exacerbation as given by O’Donnell et al5 as a guideline for the response to the question. Data were entered through a centralized, secure web-based entry system at the University of North Carolina. Spirometry pre- and post-bronchodilator results were abstracted. The diagnosis of pulmonary nontuberculous mycobacteria (NTM) was extracted from medical records and reported culture results. Sputum culture results were also reported.
The database was interrogated for data associated with a physician diagnosis of severe alpha-1 AATD, CVI and PCD. Selected characteristics included demographic and clinical characteristics, chest imaging, microbiology and pulmonary function. Severe AATD was defined as those participants having phenotype PiZZ or PiSZ. CVI was defined as those reported to have an IgG ≤ 500 mg/dl nearest to the time point of enrollment. Data on intravenous immunoglobulin (IVIG) therapy were not available. PCD was defined as those showing characteristic clinical manifestations such as organ inversus with confirmatory testing such as genetic studies, mucosal biopsy, and nasal nitric oxide as indicated by the clinical center.
The institutional review board of each participating site approved the study as well as an administrative institutional review board for the data collecting center. All participants in the BRR gave written informed consent.
Statistical Analysis
Descriptive statistics were computed for main demographic and clinical characteristics stratified by rare disease group. The results for continuous variables were reported as mean ± SD, and for categorical variables by frequencies and proportions. Values between the groups were compared using non-parametric Kruskal-Wallis test due to non-normal distribution of continuous variables, and Chi-squared/Fisher’s exact test for categorical variables. The significance level was set at 0.05. Post-hoc analyses were performed using Dunn’s test for continuous variables and using adjusted standardized residuals for categorical variables to determine the location of statistical significance. SAS 9.4 was used to perform the statistical analyses.
Results
Of the total of 2170 individuals enrolled as of January 2017, data on the status of the diagnoses of interest were available for 155 individuals with 1 diagnosis and 460 individuals, in whom these conditions had been excluded, who served as controls (termed “idiopathic”) (Figure 1). Of the 615 participants included in the present study, AATD was diagnosed in 9.4% (n=58), CVI in 2.9% (n=18), and PCD in 12.8% (n=79). Of the 58 participants reporting AATD phenotype, 37 (63.8%) were PiSZ and 21 (36.2%) were PiZZ.
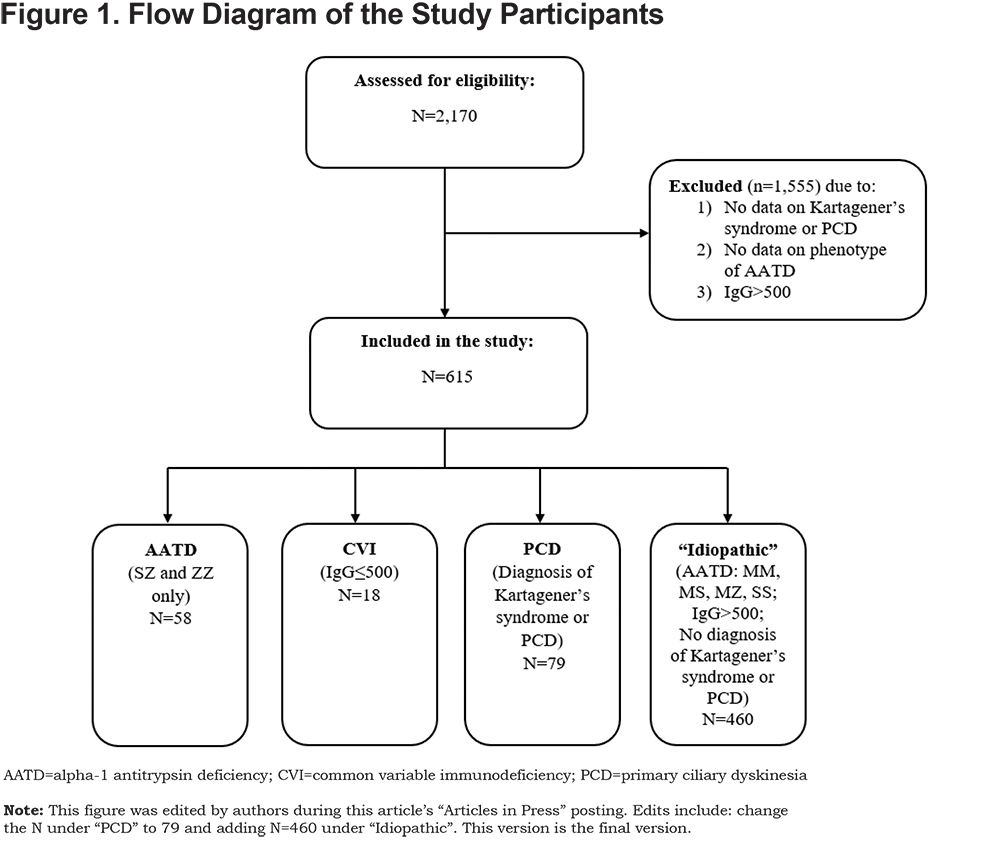
Baseline Demographics and Medical History
At enrollment, the mean age of the PCD group (41.9 ± 14.5 years) is significantly less than the mean age of the AATD (66.9 ± 10.7 years), CVI (66.7 ± 10.5 years) and control groups (64.2 ± 15.9 years) (p<.0001) (Table 1). A diagnosis of bronchiectasis was made at a much younger age in those with PCD than in the other groups (p<.0001). Table 1 also shows select baseline demographics and medical history characteristics of the study cohort. With regards to pulmonary diagnoses, COPD was more common in those with AATD. Reported pulmonary exacerbations were significantly more common in the group with PCD (p =0.002). When compared with the idiopathic group and those with AATD, a greater percentage of the group with PCD and CVI reported hospitalizations in the prior 2 years (p<.0001).
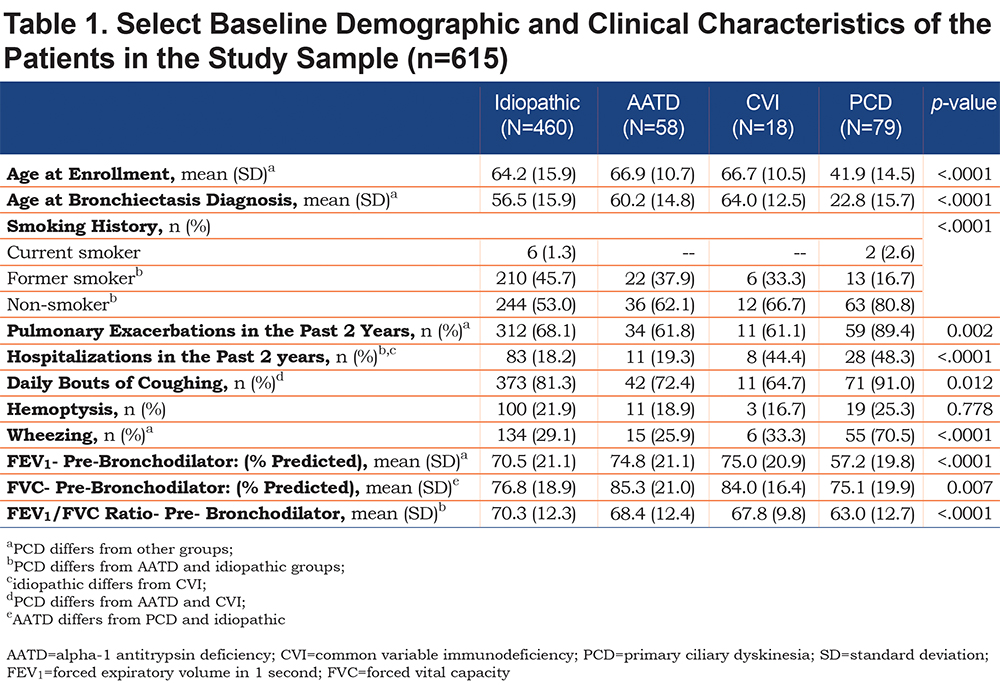
A greater percentage of participants with PCD reported daily bouts of productive cough and hemoptysis compared with the CVI and AATD groups but these symptoms were not significantly more common overall compared to the other groups. Reports of dyspnea, chest pain and fatigue were not significantly different between the groups (data not shown). Undue fatigue was reported in 46%-66% of participants. Daily bouts of coughing were significantly more common in PCD patients than those with AATD or CVI (91.0% versus 72.4% and 64.7%). Out of 19 patients with PCD who reported experiencing hemoptysis, 2 (10.5%) required bronchial embolization or surgery. None of the AATD or CVI patients who reported experiencing hemoptysis required bronchial embolization or surgery.
Pulmonary Function
Table 1 shows mean spirometry values at baseline. Spirometry was performed in 93.1% of participants with AATD, 88.2% of participants with CVI, 98.7% of participants with PCD, and 96.7% of idiopathic controls. All 3 groups showed a reduced mean forced expiratory volume in 1 second (FEV1) to forced vital capacity (FVC) ratio (< 0.7) indicating airflow obstruction and a diagnosis of COPD. As a group, participants with PCD had lower mean spirometry values with a significantly lower pre-bronchodilator FEV1 % predicted compared to the other groups. There was no statistically significant difference in % predicted FEV1 and FVC values between the study groups among participants with post bronchodilator data available (data not shown). In the PCD group, mean post bronchodilator FEV1% remained lower when compared to the other groups (65.4% [PCD] versus 77.8% [AATD], 77.1% [CVI], 73.7% [idiopathic]). Of those reporting pre- and post-bronchodilator spirometry data, there was no significant difference in the change in FEV1 or FVC % pre- versus post-bronchodilator between groups.
Mycobacterial Disease
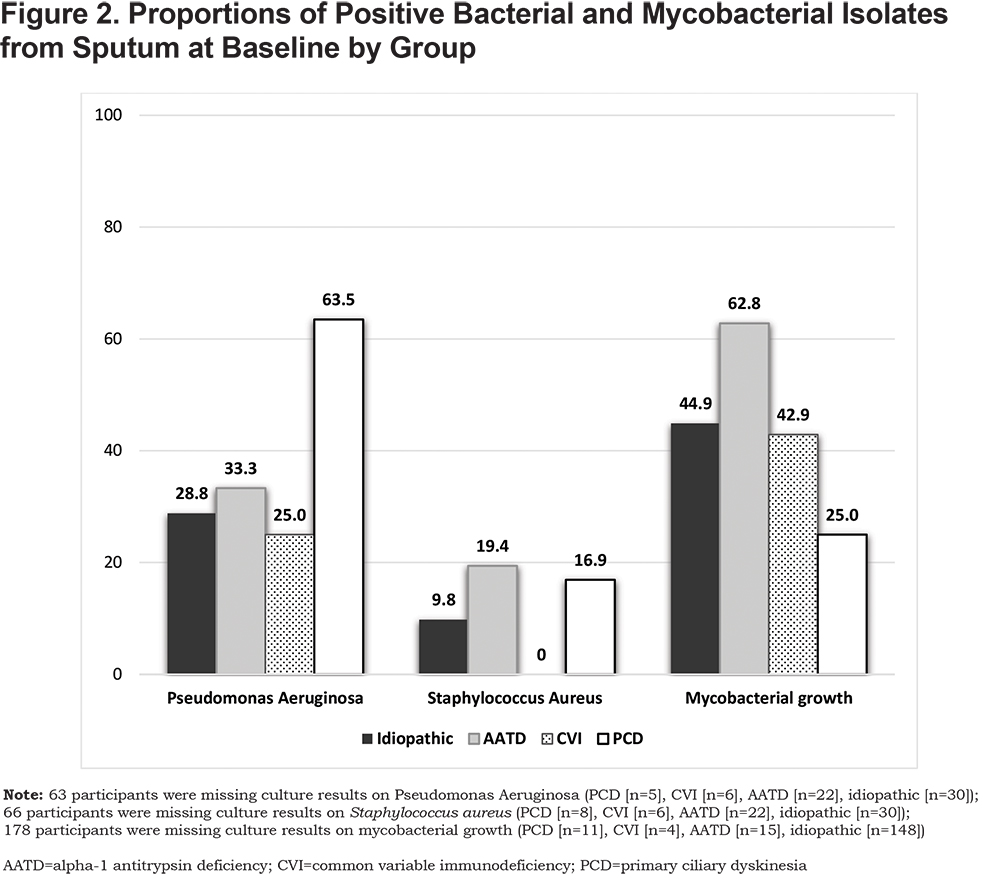
Figure 2 shows the results of mycobacterial sputum cultures. Among patients with available sputum culture results, 1 or more positive mycobacterial culture was reported in 62.8% of patients with AATD, 42.9% with CVI, 25.0% with PCD, and 44.9% with idiopathic bronchiectasis (p=0.001). M. avium complex was isolated in at least 1 of 3 cultures in 66.7% (18/27) of participants with AATD, 66.7% (4/6) with CVI and 50% (8/16) with PCD (p=0.59). M. abscessus was isolated in 3 participants (2 AATD and 1 PCD). In the idiopathic control group, M. avium complex was isolated in 75% (105/140), and M. abscessus in 22.9% (32/140).
Bacterial Culture
The registry questionnaire requested information about 3 sputum culture results. At least 1 positive bacterial culture result was reported in 129 of 155 (83%) individuals. Thirty-six (23%) individuals had missing data on all 3 cultures.
As shown in Figure 2, Pseudomonas aeruginosa and Staphylococcus aureus were the most commonly reported bacterial isolates from sputum. The percentage of PCD participants (63.5%) reported to be growing Pseudomonas aeruginosa in at least 1 sputum was significantly greater than in those with AATD (33.0%), CVI (25.0%) and the idiopathic group (28.8%) (p<.001). S. aureus was the second most common organism isolated from sputum in the PCD group (19.4%) which was methicillin-resistant in 3 cases (42.9%). In patients with AATD, S. aureus was reported in 12 of 71 (16.9%) participants of which 3 (25%) were methicillin resistant. S. aureus was not reported in patients with CVI (n=12). There was no significant difference in prevalence of other bacterial species (Haemophilus influenzae N=6, Streptococcus pneumoniae N=9, Stenotrophomonas maltophilia, N=5, Klebsiella pneumoniae N=4, Moraxella catarrhalis N=2) between groups. A positive fungal sputum culture was reported in 8 of 30 (26.7%) patients with AATD, 1 of 7 (14.3%) with CVI and 4 of 10 (40.0%) with PCD.
Discussion
Bronchiectasis is associated with numerous conditions including 3 rare but important conditions with different pathogenesis and management considerations. Severe AATD results in loss of anti-protease activity in the lung and airways. CVI results in loss of general and local protective humoral immunity, and PCD results in the lack of airway clearance from a defect in airway ciliary function. These 3 causes of bronchiectasis result, by different underlying pathophysiologic mechanisms, in local airway inflammation and a propensity to develop recurrent or chronic respiratory infection.6
This report utilizing data from the U.S. BRR compares and contrasts the features of bronchiectasis in these conditions with a control idiopathic subgroup, in which these conditions had been excluded. The BRR is a database of patients with non-cystic fibrosis bronchiectasis who were recruited from 13 institutions. This report shows the data from 615 of the original 2170 participants who had complete data available for the clinical characteristics and comparisons performed in this study. The major findings of the report are that the classic features of “wet bronchiectasis” that indicates chronic mucus hypersecretion and chronically infected sputum production occur in the group of participants with PCD more frequently and in this respect clinically resembles CF. Participants with PCD were younger and the symptoms of chronic respiratory infection and bronchiectasis led to a diagnosis of bronchiectasis at a much younger age than in either AATD or CVI or the idiopathic subgroup. As shown in Table 1, the average age at which the diagnosis of bronchiectasis is made is 40 years earlier than for either CVI or AATD and also much earlier than for participants in the BRR (mean age 54.1±17.8).1 Specifically, those with PCD have a persistent cough and sputum production, report more wheezing and hemoptysis, show a greater reduction in pulmonary function, greater morbidity from exacerbations and the presence of Pseudomonas in sputum. Bronchiectasis appears to be a more aggressive and progressive disease in these patients. This report is consistent with the prior cluster analysis of Aliberti et al7 who described chronic Pseudomonas infection to be associated with greater morbidity, more exacerbations and lower pulmonary function.
Nonproductive cough is more characteristic of patients with AATD and CVI. Persistent cough, wheezing and hemoptysis is reported less frequently and these participants as a group report fewer exacerbations. Prior reports have indicated that the diagnosis of AATD and CVI is frequently delayed.3,8 As the bronchiectasis associated with AATD and CVI is associated with less symptoms, a diagnosis of bronchiectasis may not be made until the diagnosis of the associated condition is established.
The mean age of self-reported diagnosis for the group of our patients with CVI is much greater (64±12) than is reported in the literature. In a large cohort study from Europe, most of the adults with bronchiectasis who were diagnosed with CVI were between 30-40 years of age.3 The reasons for the difference in this study are unclear although the delay in diagnosis in this study averaged 6 years. It may be that bronchiectasis is a late complication of CVI and develops as a result of recurrent pneumonia. A further suggestion is that propensity to bronchiectasis is genetically determined or related to the severity and type of immunodeficiency. For example, the multi-nation study indicates British individuals with CVI have a greater chance of diagnosis of bronchiectasis3 than those with the disease from other countries in Europe. Our small group may be selected during adulthood or have acquired the immunodeficiency later in life and may have a milder form of the disease.
Pulmonary function testing shows airflow obstruction to be a feature associated with bronchiectasis in the 3 rare disease groups. Only 41 of the 155 (28%) rare disease participants reported prior smoking suggesting the underlying pathophysiology of airflow obstruction is due to mechanisms other than exposure to smoke. Data from the 3 groups fulfill criteria for COPD compared to the idiopathic group in which mean FEV1/FVC ratio is above 70%. The mechanism for COPD in AATD is well described and investigated. Genetic susceptibility and immune factors play a role but the main underlying cause is protease/antiprotease imbalance.9 For CVI, the mechanisms for airflow obstruction have not been clearly defined, but recurrent bacterial infection is thought to play a role. For PCD the development of airflow obstruction most likely follows a similar mechanistic pathway as does cystic fibrosis where chronic mucus retention and infection damage leads to loss of airway integrity and bronchial hyper-responsiveness. Interestingly those with PCD report significantly more wheezing consistent with airway reactivity. PCD patients with Pseudomonas aeruginosa pulmonary infection show greater morbidity and worse pulmonary function at a younger age with a history of less smoking than the other groups.
In patients with available culture results, sputum bacterial profiles in the 3 rare disease groups were quite different. Bacterial cultures indicated Pseudomonas aeruginosa infection was more commonly reported in association with PCD than in the other groups in keeping with the greater reported morbidity in this condtion.7 Growth of Staphylococcus aureus in culture was the second most common bacterium reported in our cohorts with PCD and AATD in keeping with the reported findings in the BRR.10 Other bacteria such as Haemophilus were reported less commonly than in other reports.11 This is possibly a result of underreporting and/or the use of antibiotics for a clinical relapse without sputum confirmation.
As shown in Figure 2, in AATD there is a greater percentage of participants reporting NTM on sputum culture compared with other groups. This is in concordance with Kim et al12 who reported an abnormal AAT phenotype in 15% (homozygous AAT was excluded) of patients with culture proven NTM. AAT has been reported to inhibit rapidly growing mycobacterial infection in macrophages. 13 This finding raises interesting questions concerning the potential role of AATD and susceptibility to mycobacterial infection. Although in PCD, mycobacterial growth is reported less frequently compared with the other cohorts, our reported prevalence (25%) is higher for PCD than reported previously (8-11%).14 The significance of this finding is unclear but worth future studies.
This is the first study to compare and contrast the clinical features in 3 rare diseases associated with bronchiectasis, however, there are several limitations to our study including selection and reporting biases. Responses to questions and objective data were often incomplete requiring a reduction in the number of idiopathic bronchiectasis participants available for comparison. Definitions of exacerbations may have varied from center to center and there was no confirmation of uniformity in response. Therefore, results may not be extrapolated to the general bronchiectasis population as the reported information was potentially subjected to referral and reporting bias. Several major centers specializing in the diagnosis and treatment of bronchiectasis as well as the management of NTM pulmonary disease supplied information for a majority of the rare disease participants. In addition, several centers are active in research involving PCD.
Nevertheless, this report adds to the sparse body of literature concerning the clinical characteristics of bronchiectasis in these conditions.
Acknowledgements
Author contributions: EE, RC, AB, had full access to all the data in the study and take full responsibility for the integrity of the data and the accuracy of the data analysis. D A-H, TRA, CLD, LAD, AD, KF, DEG, MMJ, MRK, MLM, PGN, AEO, KNO, MAS, AS, BT, GT, GMT, KLW substantially contributed to the paper by review, comments and contribution to the dataset.
Other Contributions: The Bronchiectasis and NTM Research Registry is managed by the COPD Foundation, a 501(c)(3) nonprofit organization. The Registry is funded by the Richard H. Scarborough Bronchiectasis Research Fund, the Anna-Maria and Stephen Kellen Foundation, and the Bronchiectasis and NTM Research Registry Industry Advisory Committee (comprised of Aradigm Corp, Bayer HealthCare Inc, Grifols Inc, and Insmed Inc). Delia Prieto of the COPD Foundation and the BRR provided invaluable administrative support.
Declaration of Interest
Kenneth Olivier is supported in part by the Division of Intramural Research, National Heart, Lung and Blood Institute, National Institutes of Health. Doreen Addrizzo-Harris reports personal fees from AIT Therapeutics and Insmed. Alan Barker is supported in part by a grant provided by the COPD Foundation. Charles Daley is supported in part by a grant provided by the COPD Foundation. Leigh Anne Daniels reports personal fees from Insmed and Spark Partners as well as grants from Zambon and Parion/Vertex. Mark Metersky is supported in part by a grant provided by the COPD Foundation. Anne O’Donnell is supported in part by a grant provided by the COPD Foundation. Matthias Salathe is supported in part by a grant provided by the COPD Foundation. Byron Thomashow reports personal fees from Boehringer Ingelheim, GlaxoSmithKline, and AstraZeneca. Additionally, Byron Thomashow helped co-found the COPD Foundation and served as Board Chairman for the COPD Foundation for several years. Gregory Tino reports personal fees from Bayer, Grifols, Aradigm, and Cipla. Additionally, Gregory Tino is supported in part by a grant provided by the COPD Foundation. Kevin Winthrop reports grants and personal fees from Insmed and Bayer. All other authors have nothing to declare.