Running Head: Phase III Trials of Revefenacin in COPD Patients
Funding statement: This study was funded by Theravance Biopharma, Inc. (Dublin, Ireland). Mylan Inc., (Canonsburg, Pennsylvania) and Theravance Biopharma (South San Francisco, California) funded medical writing support.
Date of acceptance: February 22, 2019
Abbreviations: chronic obstructive pulmonary disease, COPD; forced expiratory volume in 1 second; FEV1; overall treatment effect, OTE; Global initiative for chronic Obstructive Lung Disease, GOLD; long-acting muscarinic antagonist, LAMA; long-acting beta2-agonist, LABA; inhaled corticosteroid, ICS; adverse event, AE; Medical Dictionary for Regulatory Activities, MedDRA; intention-to-treat, ITT; revefanacin, REV; standard deviation, SD; body mass index, BMI; forced vital capacity, FVC; modified Medical Research Council dyspnea scale, mMRC; COPD Assessment Test, CAT; least squares, LS; standard error, SE; confidence interval, CI; serious adverse event, SAE; gastrointestinal, GI
Citation: Ferguson GT, Feldman G, Pudi KK, et al. Improvements in lung function with nebulized revefenacin in the treatment of patients with moderate to very severe COPD: results from two replicate phase III clinical trials. Chronic Obstr Pulm Dis. 2019; 6(2): 154-165. doi: http://doi.org/10.15326/jcopdf.6.2.2018.0152
Online Supplemental Material: Read Online Supplemental Material (376KB)
Introduction
Chronic obstructive pulmonary disease (COPD) is a common and treatable disease characterized by airflow limitation and respiratory symptoms.1 The Global initiative for chronic Obstructive Lung Disease (GOLD) consensus report recommends the use of long-acting muscarinic antagonist (LAMA) and/or long-acting beta2-agonist (LABA) bronchodilators as first-line therapy for patients with persistent COPD symptoms.1 Long-acting inhaled bronchodilators are most often self-administered with hand-held devices such as pressurized metered-dose inhalers or dry powder inhalers2,3; however, many patients are challenged by the use of these inhalers (typically due to impaired hand/breath coordination, insufficient inspiratory flow rates or cognitive impairment).4,5 These patients may benefit from delivery of their medications via a nebulizer, which may provide improved symptom control and quality of life compared with other modes of delivery.6 Current standard battery-powered jet nebulizers are portable, with nearly silent operation, making them more practical for everyday use than previous nebulizers.4,6
Revefenacin is a novel, once-daily, lung-selective LAMA in late-stage clinical development for the treatment of patients with COPD.7,8 It is designed to produce sustained and localized dilation in the bronchi with minimal systemic drug exposure.9,10 In a 4-week Phase II placebo-controlled dose-finding study in patients with COPD, once-daily revefenacin (88 μg, 175 μg and 350 μg) resulted in significant improvements in trough forced expiratory volume in 1 second (FEV1).11 Here we report the results of 2 replicate 12-week Phase III trials that characterize the efficacy, safety and tolerability of once-daily revefenacin (88 µg and 175 µg) administered by standard jet nebulizer in patients with moderate to very severe COPD.
Methods
Study Design and Conduct
Study 0126 (NCT02459080) and Study 0127 (NCT02512510) were Phase III, randomized, double-blind, placebo-controlled, multiple-dose, parallel-group studies. Both studies were conducted in accordance with the principles of the International Council on Harmonisation of Technical Requirements for Pharmaceuticals for Human Use guideline for good clinical practice,12 and the code of ethics of the World Medical Association’s Declaration of Helsinki,13 and all patients provided written informed consent. Entry criteria, study design and statistical analyses for both studies were identical. The protocol was reviewed and approved by an institutional review board (Quorum Review IRB, Seattle, Washington).
Patients and Treatments
Patients were ≥ 40 years old with documented COPD history, a smoking history of at least 10 pack years, a post-ipratropium FEV1/forced vital capacity ratio of < 0.7 and a post-ipratropium FEV1 of < 80% of predicted normal but at least 700 mL at Visit 1B (online data supplement e-Figure 1), constituting criteria for moderate to very severe COPD. Patients were excluded if they had a history of myocardial infarction or unstable angina within the previous 6 months, unstable or life-threatening cardiac arrhythmia requiring intervention within the previous 3 months, New York Heart Association Class IV14 heart failure prior to the start of the study or exhibited an abnormal and clinically significant 12-lead electrocardiogram finding at study entry. If a patient did not meet the eligibility criteria because of a failed screening test, this test or procedure was not permitted to be repeated and the individual was considered ineligible. Failed screening tests resulting in ineligibility included spirometry (i.e., if an individual failed to meet any spirometry-related criteria after the first attempt, the individual was screen failed). Spirometry was assessed during the treatment period at days 1, 15, 29, 57, 84 and 85, which corresponded to Visits 3, 4, 5, 6, 7, and 8, respectively. A telephone follow-up visit was carried out approximately a week after Visit 8 to review concomitant medications and adverse events (online data supplement e-Figure 1). Patients were randomized 1:1:1 to receive revefenacin 88 μg, revefenacin 175 μg or matching placebo, administered once daily in the morning by a standard jet nebulizer (PARI LC® Sprint, Starnberg, Germany) for 12 weeks. Concomitant LABA-containing therapy (with or without inhaled corticosteroids [ICSs]) was permitted in up to 40% of the study population to ensure robust assessments of concurrent therapies used by the participants. Once the 40% limit was reached, new individuals who entered screening required a 14-day washout of any LABA-containing therapy prior to the ipratropium reversibility test at screening. Individuals on ICS/LABA combination therapy enrolled after the 40% cap was reached had their medication modified to receive only ICS monotherapy at an equivalent dose for at least 14 days, prior to the ipratropium reversibility visit at screening. Stable doses of ICSs without concomitant LABAs were permitted but use of LAMAs and short-acting muscarinic antagonists was prohibited.
Assessments and Endpoints
The primary efficacy endpoint was change from baseline in trough FEV1 (defined as the mean of the 23.25- and 23.75-hour spirometry assessments following the 84th dose) on day 85. Secondary efficacy endpoints included overall treatment effect (OTE) on trough FEV1 (defined as the inverse-variance weighted average of all the trough FEV1 assessments across days 15 through 85 [15, 29, 57 and 85]) and peak (maximum) FEV1 (0-2 hours post first dose) on day 1. Patient-reported outcomes, which will be reported elsewhere, included rescue albuterol use, the St George’s Respiratory Questionnaire,15 and the Transition Dyspnea Index.16 Additional details on secondary efficacy endpoints are described in the online data supplement.
Safety assessments included treatment-emergent adverse events (AEs), clinical laboratory measurements, electrocardiograms (reported in a separate paper), vital signs and physical examinations. AEs were coded using preferred terms from the Medical Dictionary for Regulatory Activities (MedDRA®), Version 18.1.17 System organ class, preferred term, relationship to study drug, and severity summarized the frequency and percentage of patients reporting AEs.
Statistical Analysis
The intention-to-treat (ITT) analysis set included all randomized patients who received at least 1 dose of the study drug (revefenacin or placebo) and had at least 1 recorded post-baseline FEV1 assessment. Assessments of efficacy were performed using the ITT analysis set. Assessments of safety were performed using the safety analysis set, which included all randomized patients who received at least 1 dose of study drug. Additional statistical analyses are described in the online data supplement.
Results
Patients
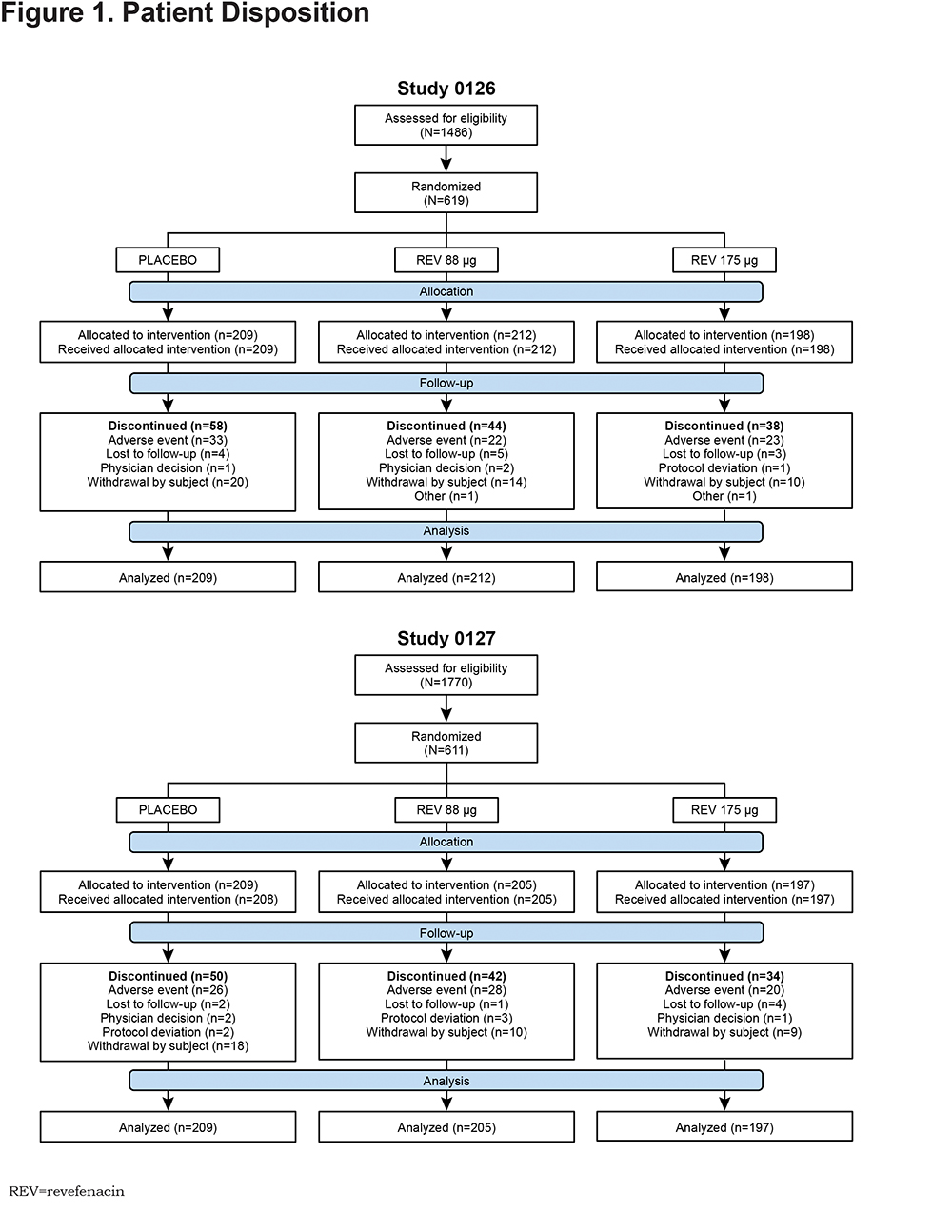
A total of 3256 patients were screened for both studies (Figure 1), of which 619 were randomized to treatment in Study 0126 (revefenacin 88 μg [n=212], revefenacin 175 μg [n=198] and placebo [n=209]) and 611 were randomized to treatment in Study 0127 (revefenacin 88 μg [n=205], revefenacin 175 μg [n=197] and placebo [n=209]). One patient randomized to placebo in Study 0127 never received study treatment and was therefore excluded from the final ITT analysis set. Thus, 610 patients contributed efficacy data and were included in the ITT analysis set. Another patient randomized to the revefenacin 175 μg group in Study 0127 received the wrong study drug kit at Visit 6 (day 57) and was subsequently dosed with placebo. This patient thus contributed safety data to 2 treatment groups (175 μg and placebo) and, therefore, 611 patients are presented in the Study 0127 safety analysis set. The ITT and safety analysis sets for Study 0126 included 619 patients in both cases.
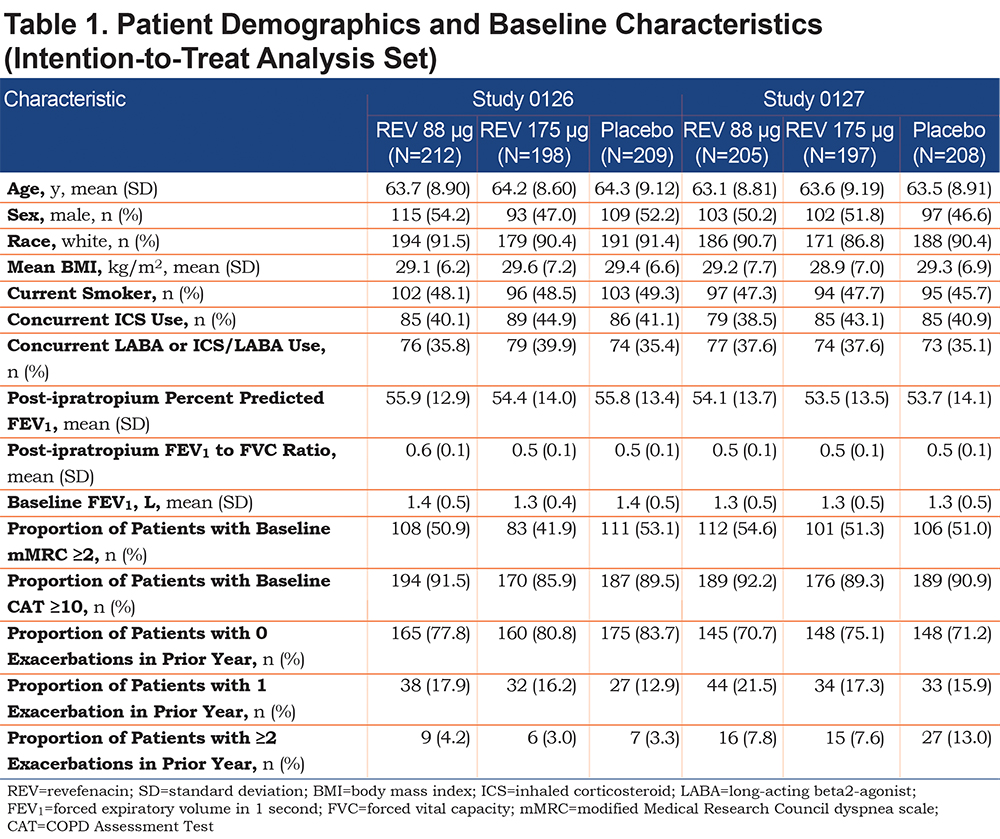
Patient demographics and baseline characteristics from Studies 0126 and 0127 (Table 1)indicated revefenacin and placebo groups were well balanced across all variables, including demographic background, concurrent LABA and/or ICS use and pulmonary health status. Baseline COPD severity of patients (online data supplement e-Figure 2) demonstrated similar rates of severe airflow limitation (GOLD 3) and very severe airflow limitation (GOLD 4)18 in Study 0126 (29.1% and 4.5%, respectively) and Study 0127 (34.9% and 4.1%, respectively). Similar rates of patients with more symptoms and high risk of exacerbations (2011 GOLD category D18) were observed between the studies (Study 0126 [31.5%] and Study 0127 [36.4%]).
Efficacy
Primary Endpoint
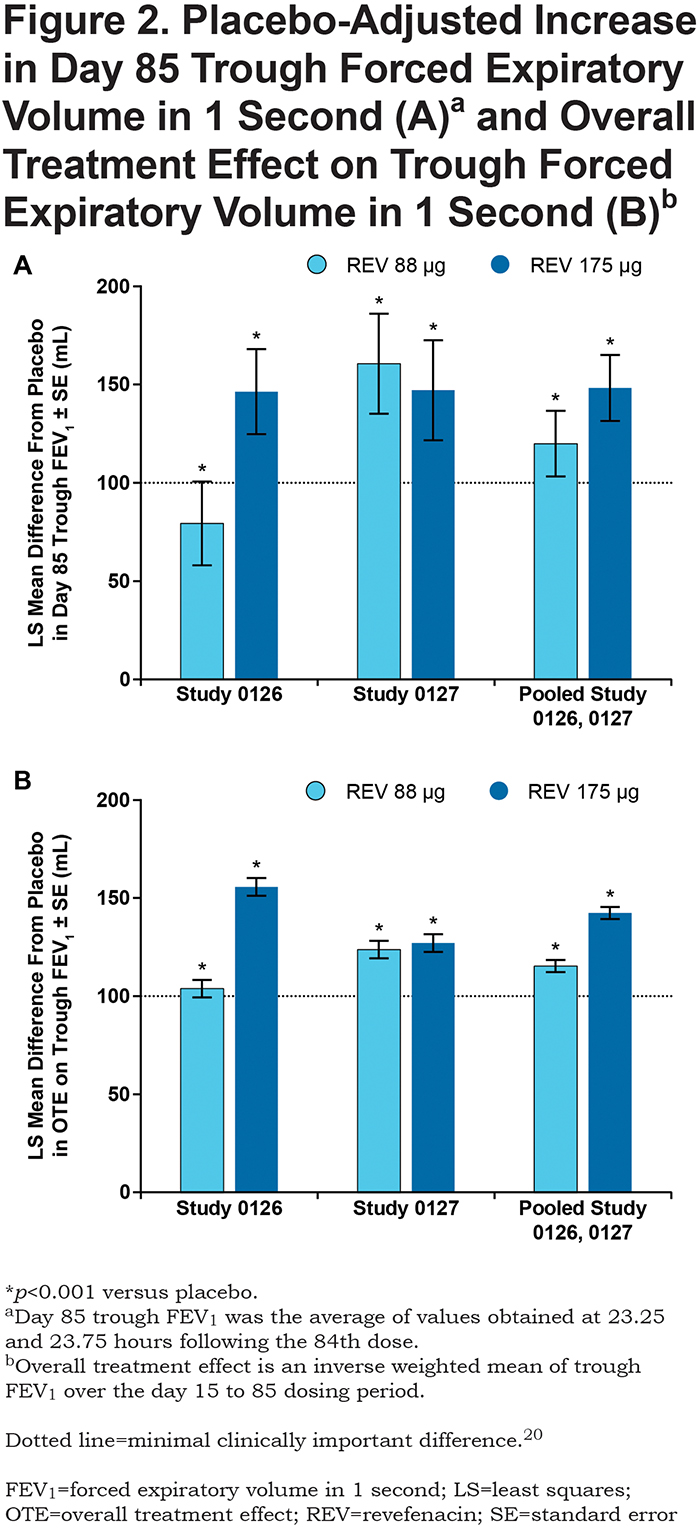
Revefenacin (88 µg and 175 µg) improved day 85 baseline-adjusted mean trough FEV1 compared with placebo in both Study 0126 and Study 0127 (Figure 2A). In Study 0126, the placebo-adjusted least squares (LS) mean increase in trough FEV1 was 79.2 mL for revefenacin 88 µg (p=0.0003) and 146.3 mL for revefenacin 175 µg (p<0.0001). In Study 0127, the LS mean increase in trough FEV1 with revefenacin was 160.5 mL (88 µg) and 147.0 mL (175 µg) (both p<0.0001). Analysis of pooled Study 0126 and Study 0127 results revealed placebo-adjusted increases in trough FEV1 of 119.8 mL (88 µg) and 148.1 mL (175 µg), respectively; the 28.3 mL difference in trough FEV1 between the revefenacin 88 µg and 175 µg doses was not statistically significant (p=0.088).
Secondary Endpoints
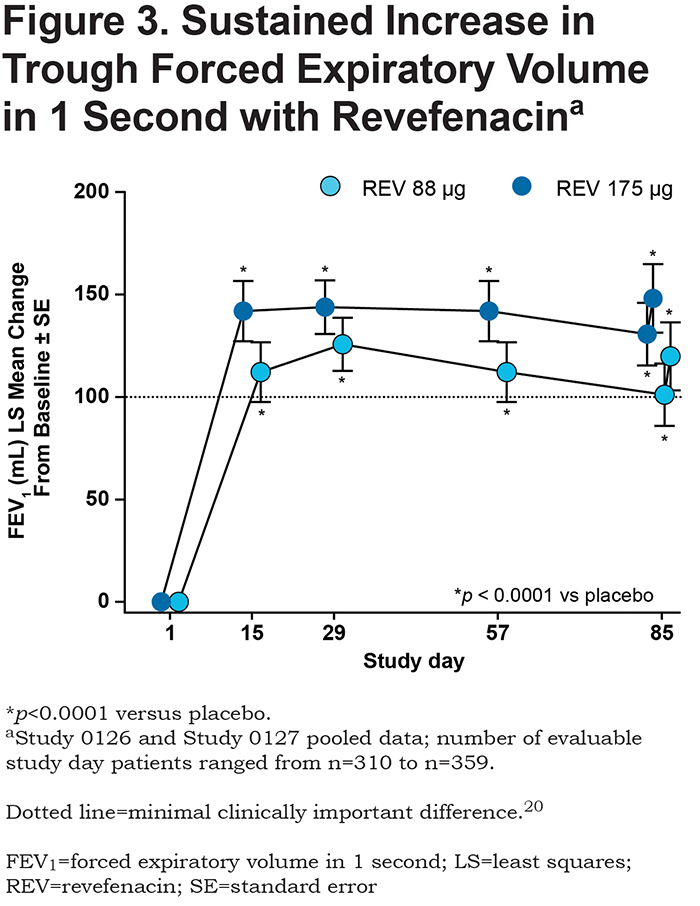
Revefenacin increased OTE trough FEV1 by ≥ 100 mL compared with placebo in both Study 0126 and Study 0127 (Figure 2B). Analysis of pooled Study 0126 and Study 0127 results revealed placebo-adjusted increases in OTE FEV1 of 115.3 mL (88 µg) and 142.3 mL (175 µg). In addition, revefenacin increased trough FEV1 by ≥ 100 mL on days 15, 29, 57, 84 and 85 versus placebo at both the 88 µg and 175 µg dose (Figure 3).
A significant increase in FEV1 occurred within 2 hours of the first treatment with revefenacin in both studies. Analysis of pooled study results revealed placebo-adjusted LS mean increases in peak FEV1 (0-2 hours after first dose) of 127.3 mL (88 µg) and 129.5 mL (175 µg) (both p<0.0001).
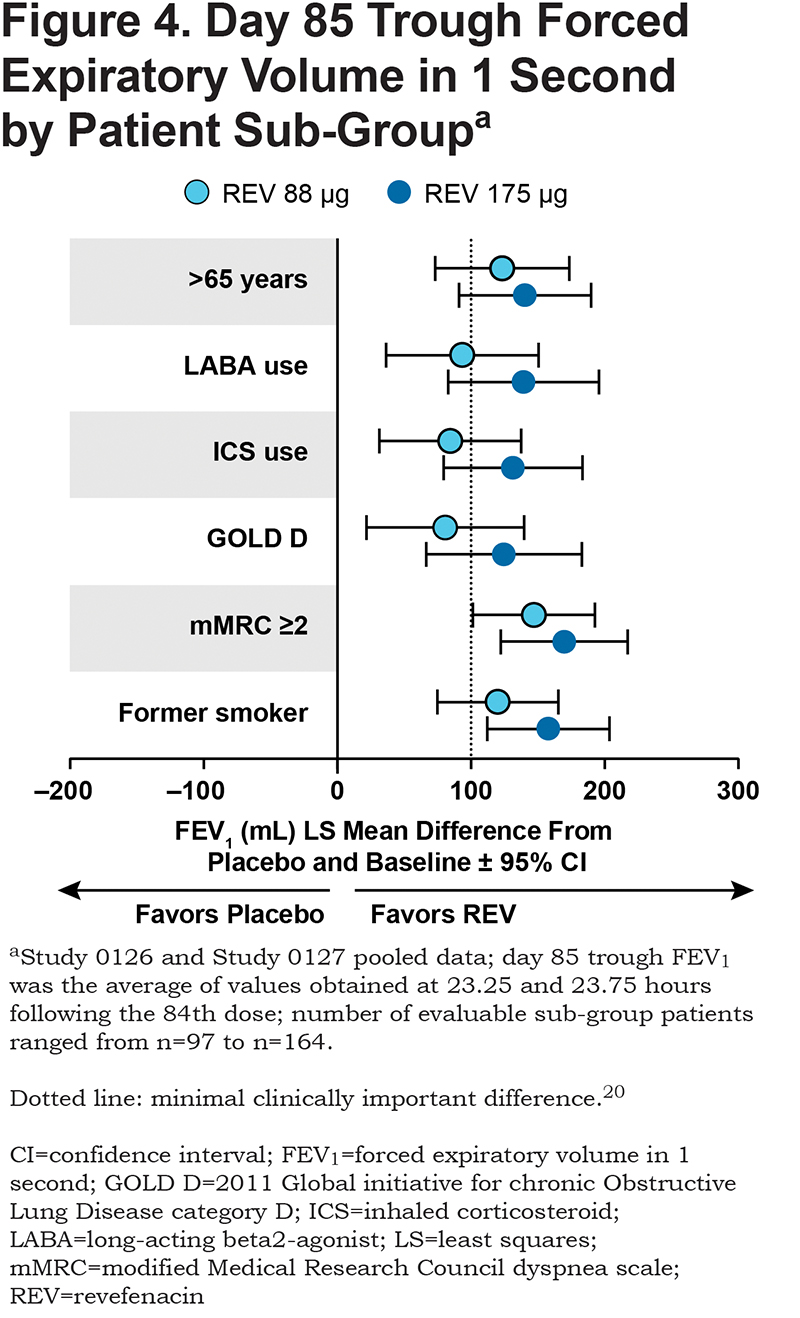
Patient Sub-group Analysis
Analysis of pooled Study 0126 and Study 0127 results showed revefenacin 175 µg produced greater improvements in day 85 trough FEV1 than revefenacin 88 µg. The patients in the 175 μg group likely had more severe COPD, because this group included those taking concomitant LABA therapy along with ICSs, those > 65 years of age, in 2011 GOLD category D,1817 with a modified Medical Research Council dyspnea scale ≥ 2 and former smokers. Nevertheless, both doses were significantly superior to placebo (Figure 4).
Safety
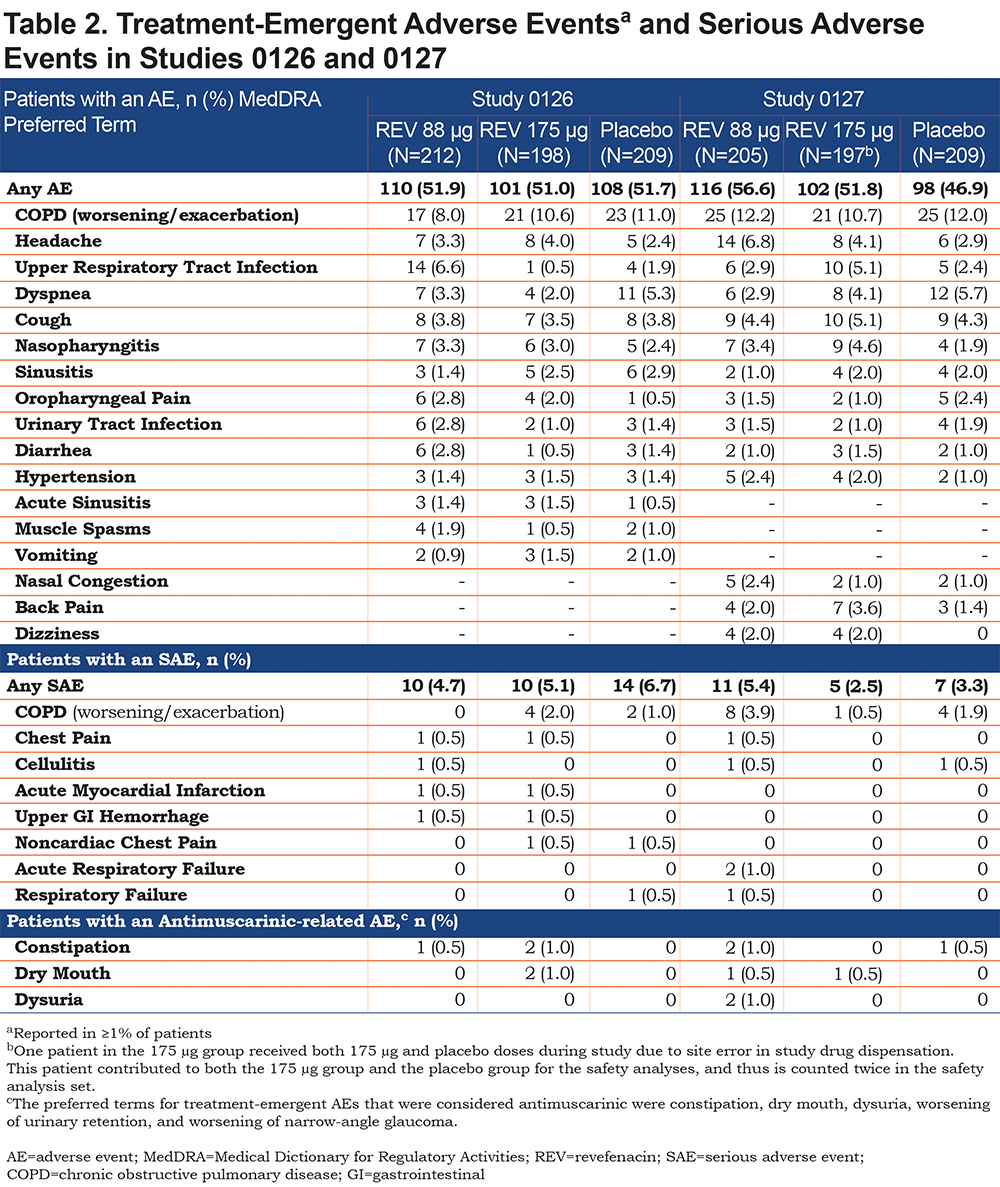
The overall incidence of treatment-emergent AEs was similar in the revefenacin (88 µg and 175 µg) and placebo treatment groups for Study 0126 and Study 0127 (Table 2). Approximately 47% to 57% of patients by treatment group reported at least 1 AE. COPD (worsening/exacerbation) was the highest-incidence AE (≤ 12.2%). Headache (≤ 6.8%), respiratory infection (≤ 6.6%), dyspnea (≤ 5.7%) and cough (≤ 5.1%) were the next most common AEs, with similar frequencies between treatment groups.
Antimuscarinic-related AEs were both infrequent and evenly distributed between treatment groups, including placebo, in both studies, with no patient in either study experiencing more than 1 antimuscarinic AE. The most common antimuscarinic AEs were constipation and dry mouth. A total of 6 patients, between both studies, reported constipation (Study 0126: 1 in the revefenacin 88 µg group and 2 in the 175 μg group; Study 0127: 1 in the placebo group, 2 in the 88 µg group) and dry mouth was reported in 4 patients (Study 0126: 2 patients in the 175 μg group; Study 0127: 1 patient each in the 88 μg and 175 μg groups).
The overall incidence of serious AEs (SAEs) was similar for revefenacin and placebo in Study 0126 (≤ 6.7%) and Study 0127 (≤ 5.4%) (Table 2). In Study 0126, 1 SAE, a case of worsening/exacerbation of COPD in the 175 μg group, was considered by the investigator to be treatment related, while in Study 0127, 1 SAE of pneumonia in the revefenacin 175 µg group was considered to be related to the study drug; no other SAEs were considered study-drug related.
Five events over the course of both Study 0126 and Study 0127 met major adverse event criteria. In Study 0126, 1 cardiovascular death occurred in the placebo group, 1 myocardial infarction/unstable angina event occurred in both the 88 μg and 175 μg groups and 1 arrhythmia event occurred in the 88 μg group. In Study 0127, 1 non-cardiovascular death (homicide) occurred in the 175 μg group. A pre-specified adjudication of all major adverse cardiac adverse events in the Phase III program by an independent external clinical events committee (the Duke Clinical Research Institute) determined that none of the cardiovascular events was related to the study drug.
Discussion
Studies 0126 and 0127 both met their primary endpoint of increased trough FEV1 compared with placebo after 12 weeks of once-daily revefenacin in patients with moderate to very severe COPD and in those receiving concomitant LABA therapy. The inclusion of patients with very severe COPD was unique to this monotherapy program as previous studies with revefenacin were conducted in patients with moderate-to-severe COPD.11,19 Both dose levels of revefenacin showed clinically meaningful improvements in trough FEV1 over the entire treatment period, while the revefenacin 175 µg dose delivered additional benefit over the 88 µg dose in the ITT population as well as in key sub-groups, such as patients with markers of more severe disease. For Study 0126, trough FEV1 technically did not meet the 100 mL minimal clinically important difference level20; however, 40% of these patients were on a LABA, and minimal clinically important difference only pertains to a comparison with placebo, so the degree of FEV1 improvement would be expected to be lower. The efficacy results from the 2 studies were consistent with previous studies of revefenacin, including a 4-week Phase II trial in which revefenacin demonstrated significant improvements in trough FEV1 compared with placebo.11,19 Moreover, the trough FEV1 improvements with the 88 µg and 175 µg dose (119 mL and 148 mL, respectively [pooled study results]) are comparable with reported values for another long-acting bronchodilator, tiotropium, approved for the treatment of COPD.21
While the 88 μg and 175 μg doses met the pre-specified primary endpoint in both studies, there were differences in response to the 88 μg dose between the 2 studies, though there were no clear differences in baseline characteristics between them. One possibility for the differences is that the lower dose was at the lower edge of the dose-response curve and some patients in the one study might not have received the full benefit of the drug, which was corrected by the higher dose. This suggests that the higher dose would be the more optimal dose for all participants, especially as there were no added safety concerns with the higher dose. In addition, the LS mean difference from placebo in trough FEV1 at Day 85 was slightly higher for the 88 µg dose than the 175 µg dose in Study 0127. This result was a single time point. In the pooled results (studies 0126 and 0127), the LS mean difference for placebo in trough FEV1 at Day 85 was higher for the revefenacin 175 µg dose than for the revefenacin 88 µg dose. Therefore, it is possible that there is an additional response with the revefenacin 175 µg dose.
Revefenacin also demonstrated a desirable safety and tolerability profile for both doses in both studies, which was consistent with previous studies of revefenacin in patients with COPD.11,19 In addition, a low incidence of antimuscarinic AEs was observed. This finding is consistent with revefenacin’s pharmacologic properties of competitive antagonism of the M3 receptor, unique molecular class (i.e., it is not a quaternary ammonia) and lung-selective design,7,8 as well as with results from previous clinical studies.11,19 The safety profile was supported by a prior pre-clinical study8 that revealed revefenacin’s superior functional lung selectivity index (ratio of bronchoprotective versus antisialagogue potency) compared with either glycopyrronium or tiotropium, with dose-dependent, 24-hour bronchoprotection that was maintained after 7 days of once-daily dosing in animal models of bronchoconstriction. Pooled results from Study 0126 and Study 0127 were similar to each individual study with regard to efficacy and safety outcomes.
Limitations of the replicate studies include the treatment period being only approximately 3 months, which does not allow for conclusions regarding long-term treatment. Although study patients were provided with rescue medication that could potentially impact outcomes, reductions for both revefenacin doses in the average number of rescue medication puffs per day used, while not statistically significant, showed trends in favor of revefenacin over placebo and supported the results of the primary endpoint (data not shown). Results from a 52-week safety study of revefenacin in COPD (to be published separately) should help elucidate revefenacin’s long-term safety profile. Additionally, the subgroup analysis results should be interpreted with caution due to the smaller sample sizes in each subgroup.
A key strength of the studies, apart from their blinded and controlled design, is that they were replicate studies that observed similar results for both primary and secondary endpoints, thereby adding consistency and validity to their outcomes. Furthermore, compared with placebo, both revefenacin doses in the pooled analysis increased trough FEV1 by >100 mL, a level of bronchodilation that has previously been suggested as a minimally clinically important difference for FEV1.20
As a novel once-daily LAMA for nebulization, revefenacin may be differentiated from the currently available once-daily tiotropium and umeclidinium handheld products in important ways. Revefenacin’s novel biphenyl carbamate tertiatry amine structure is distinct from the quaternary ammonium antagonists (e.g., tiotropium and umeclidinium),22 thus representing the first inhaled LAMA of its class in clinical development for COPD. Revefenacin is a LAMA endowed with chemical stability (enabling long-term storage as a preservative-free aqueous solution product), high lung-to-salivary gland functional selectivity (vida infra) and a metabolically labile primary amide “soft-drug” site to allow rapid systemic clearance of the parent drug, thus potentially minimizing systemically mediated AEs.7
While the high metabolic lability of revefenacin contrasts with the relative metabolic stability of tiotropium and its primarily renal systemic clearance profile, revefenacin shares umeclidinium’s profile of rapid metabolic turnover after distributing from the lung.23 Systemic clearance of revefenacin is primarily via enzymatic hydrolysis (versus cytochrome P450 [CYP2D6]-mediated oxidative turnover for umeclidinium).24 Finally, the revefenacin drug product in the current study was administered via a standard jet nebulizer, and this unique presentation of a once-daily bronchodilator may be of future therapeutic benefit to those patients who prefer or require nebulization therapy.4
Conclusion
Revefenacin (88 µg and 175 µg), administered once daily for 12 weeks to patients with moderate to very severe COPD, demonstrated clinically significant improvements in trough FEV1, as well as OTE FEV1, over the entire treatment period. Revefenacin 175 µg demonstrated greater improvements in FEV1 in concomitant LABA patients and in more severe patients than revefenacin 88 µg. Revefenacin has the potential to be the first once-daily, long-acting bronchodilator for use in patients who require or prefer nebulized antimuscarinic therapy.
Acknowledgments
This study was funded by Theravance Biopharma R&D, Inc., (Dublin, Ireland). The authors acknowledge Roger Hill, PhD, and Gautam Bijur, PhD, for medical writing and Paula Stuckart for editorial assistance in the preparation of the manuscript (Ashfield Healthcare Communications, Middletown, Connecticut).
Author contributions: GTF takes responsibility for the content of this article, including the data and analysis. GTF, GF, KKP participated in data acquisition and data analysis/interpretation.
CNB, EJM, BH, SP, GC participated in study design, data acquisition and data analysis/interpretation. All authors contributed to manuscript drafting and/or critical revision, approved the final manuscript and agree to be held accountable for all aspects of the work.
Declaration of Interest
GTF has received grants, served on advisory boards, received consulting fees and/or received speaking fees from AstraZeneca, Boehringer Ingelheim, Circassia, Forest, GlaxoSmithKline, Innoviva, Meda, Mylan, Novartis, Pearl Therapeutics, Sanofi, Sunovion, Theravance Biopharma and Verona. GF and KKP have nothing to declare. CNB, EJM, BH, SP and GC are employees of Theravance Biopharma US, Inc. Mylan Inc., (Canonsburg, Pennsylvania) and Theravance Biopharma US, Inc., (South San Francisco, California) funded medical writing support.