Running Head: Alpha-1 and Pulmonary Langerhans Cell Histiocytosis?
Funding Support: None
Date of Acceptance: April 8, 2019
Abbreviations: pulmonary Langerhans cell histiocytosis, PLCH; alpha-1 antitrypsin, AAT; protease inhibitor, Pi; alpha-1 antitrypsin deficiency, AATD; Langerhans cells, LC; mitogen activating protein kinase, MAPK
Citation: Ellis PR, Campbell EJ, Turner AM, Stockley RA. Alpha-1 antitrypsin deficiency: a predisposing factor for the development of pulmonary Langerhans cell histiocytosis? Chronic Obstr Pulm Dis. 2019; 6(3): 206-209. doi: http://doi.org/10.15326/jcopdf.6.3.2019.0129
Case Report
The coexistence of 2 rare diseases raises the possibility that their underlying pathophysiology is related. Single instances of such cases may provide new insights into otherwise poorly understood disease mechanisms. We describe the case of a young Asian male diagnosed with pulmonary Langerhans cell histiocytosis (PLCH) who was also found to have an exceptionally rare genotype resulting in absence of circulating alpha-1 antitrypsin (AAT).
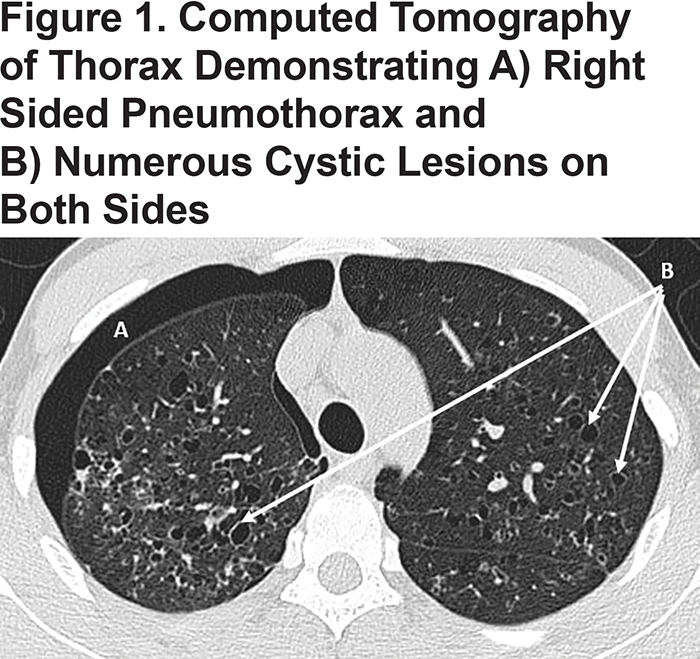
The 22-year-old cigarette smoker presented with a refractory spontaneous pneumothorax. Computed tomography of the thorax confirmed numerous pulmonary cystic lesions, especially in the upper and middle zones (Figure 1). Following surgical resolution of the pneumothorax, PLCH was confirmed by lung biopsy; numerous eosinophilic inflammatory infiltrates in association with Langerhans cells were seen that stained positively for CD1A, S100 and langerin. The patient also had a strong family history of lung disease. During admission, an AAT level was requested and found to be undetectable. Genetic analysis revealed an unprecedented homozygous status for a rare null variant, protease inhibitor (Pi)*Q0saarbruecken.
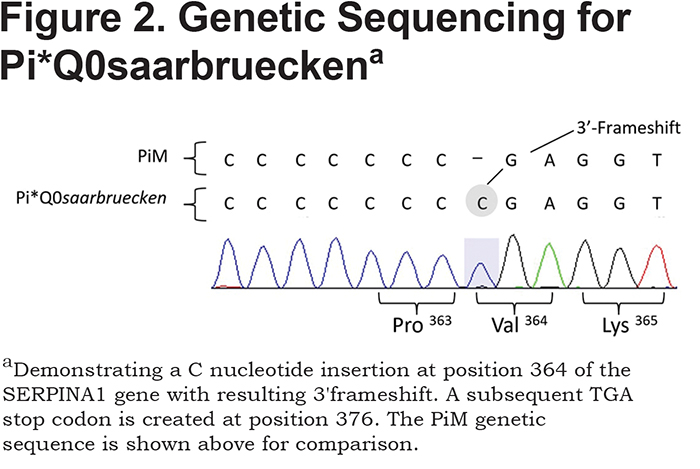
AAT deficiency (AATD) is associated with an increased inflammatory response in lung tissue and reduced inhibition of neutrophil proteinases perpetuates this process. Over 500 genetic variations of AAT exist, with the most common deficiency states being the SZ and ZZ genotypes that have serum AAT levels approximately 25% and 15% of normal, respectively. Our case was homozygous for Pi*Q0saarbruecken, a null variant of AATD resulting in <0.1% of normal serum AAT. The Pi*Q0saarbrueken allele arises from a single C-nucleotide repeat insertion in exon 5 of the SERPINA1 gene on chromosome 14 (Figure 2). The resulting 3' frameshift leads to a premature stop codon at position 376.1 The truncated glycoprotein therefore lacks the essential 391 proline residue that enables AAT transportation out of the hepatocyte and into the circulation.2 The Pi*Q0saarbruecken allele has been described twice in the literature; Faber et al2 identified 2 healthy heterozygotes from the same family and Lin et al1 described an individual with 2 Z alleles, one of which also contained the Pi*Q0saarbruecken mutation. The relationship of null variants to pulmonary inflammation is unknown, although such patients have worse lung function than ZZ deficient individuals.
PLCH is a diffuse lung disease that is more common in young smokers and that varies in severity from self-limiting to life threatening. A uniform pathologic feature of PLCH is the presence of airway inflammation.3 In PLCH, CD1A positive dendritic cells known as Langerhans cells (LCs) accumulate in clusters adjacent to bronchiolar airways together with other immune cells including lymphocytes, macrophages and eosinophils. These complex inflammatory granulomas are thought to be responsible for subsequent destruction and remodelling of airways, producing numerous cystic lesions as seen in the current case (Figure 1). LCs present in PLCH granulomas function poorly as antigen presenting cells, suggesting that tissue destruction may not occur via direct cytotoxic effects of T lymphocytes.4 Other causes of tissue destruction have been proposed, including activity of metalloproteinases that degrade extracellular matrix proteins,5 and tissue injury from poorly inhibited serine proteinase could also play a role.
There is emerging evidence that a neoplastic process can contribute to the initial development of PLCH in some cases. Mutations in signalling molecules that increase cell survival and proliferation via the mitogen activating protein kinase (MAPK) pathway have been identified in systemic and pulmonary forms of Langerhans cell histiocytosis. The most commonly associated mutation, BRAFV600E, was found in 28% of patients with PLCH in one study.6 The clinical importance of BRAFV600E and other mutations associated with PLCH are yet to be fully characterized, although targeted therapies are currently being assessed in particularly aggressive cases.
Regardless of the contribution of genetic susceptibility, there is a striking relationship between the risk of PLCH and a current or previous smoking history.7 Cigarette smoke may contribute to the development of PLCH through several mechanisms: (1) induction of immune cells to release cytokines that facilitate activation and maturation of LCs; (2) enhanced survival of LCs through mechanisms that oppose apoptosis, and (3) increased levels of chemoattractant that recruit LCs.8 Cases may also regress spontaneously following smoking cessation, further suggesting that smoking is integral to the development and persistence of the disease. We note that serine proteinases have also been implicated in both cigarette-driven inflammation and malignant proliferation,9 and that absence of a major inhibitor of serine proteinases in our case could increase susceptibility to airway injury.
In the case described it seems highly unlikely that 2 such rare pulmonary diseases occurred independently. It is possible that development of PLCH was driven by a proinflammatory environment created by cigarette smoking in the complete absence of AAT. Metalloproteinases, which are activated by serum proteinases such as neutrophil elastase, have been implicated in PLCH5 although the pathway has not been clarified. Serine proteinases may have an indirect effect on the pathophysiology of PLHC exaggerated by the complete absence of AAT.
With regard to our case, we note the precedent that other rare diseases characterized by tissue and vascular inflammation, i.e., necrotizing panniculitis and granulomatosis with polyangiitis, have been causally linked to AATD.10 It is particularly relevant that augmentation therapy can be highly effective with necrotising panniculitis associated with AATD. We believe it may be appropriate to test for AATD in cases of PLCH, to explore the relationship between these 2 disease entities further, and to consider augmentation therapy for severe and/or refractory cases of PLCH, particularly in individuals who have AATD.
Acknowledgments
PRE, AMT and RAS contributed to the initial concept. EJC characterized the AAT mutation, PRE collected data and prepared the initial manuscript. PRE, EJC and RAS reviewed and made significant contribution to the final manuscript.
Declaration of Interest
All declarations of interest fall outside the submitted work. RAS reports grants and personal fees from CSL Behring, Boehringer, Astra Zeneca, Mereobiopharma, Shire, Chiesi, and Akari. AMT reports grants from Grifols Biotherapeutics and the Alpha-1 Foundation, personal fees from CSL Behring and grants and non-financial support from Arrowhead Inc. EJC reports personal fees from HerediLab, Inc. PRE declares no conflicts of interest.