Running Head: Alpha-1 Antitrypsin Deficiency and Liver Disease
Funding support: not applicable
Date of Acceptance: December 11, 2019
Abbreviations: alpha-1 antitrypsin deficiency, AATD; alpha-1 antitrypsin, AAT; endoplasmic reticulum, ER; ER-associated degradation, ERAD; ubiquitin-proteasome system, UPS; inclusion bodies, IBs; periodic acid–Schiff, PAS; unfolded protein response, UPR; body mass index, BMI; hepatocellular carcinoma, HCC; heat shock response, HSR; jun-N-terminal kinase, JNK; heptacyte nuclear factor 4 alpha, HNF-4α
Citation: Bouchecareilh M. Alpha-1 antitrypsin deficiency-mediated liver toxicity: why do some patients do poorly? What do we know so far? Chronic Obstr Pulm Dis. 2020; 7(3): 172-181. doi: http://doi.org/10.15326/jcopdf.7.3.2019.0148
AATD-Mediated Liver Disease: General Introduction
Alpha-1 antitrypsin deficiency (AATD), is a rare, inherited disorder that affects approximately 1 in 2000-5000 births in the white population1 and is associated with lung (emphysema) and/or liver damage (cirrhosis). Mutations in the SERPINA1 gene that encodes the alpha-1 antitrypsin (AAT) protein cause this deficiency, leading to a reduced level of AAT in serum.2,3 The normal SERPINA1 allele is referred to as M allele and more than 150 SERPINA1 mutants have been described to date. Nevertheless, only a few of these mutants mediate liver disease. Among all those variants, the Z allele is the most frequent variant associated with liver disease.4 It is caused by a point mutation, a single amino acid substitution (Glu342Lys), which results in an aberrantly folded protein.1,5 Unlike the wild-type (WT) AAT protein secreted by the hepatocytes (primary and major site of production of AAT), the Z mutant folds inefficiently and never reaches the trafficking and secretion pathway. Consequently, homozygous ZZ patients have approximately only a 10%-15% circulating level of AAT compared to the WT.
The Z variant mutation leads to the retention of the Z protein within the hepatocyte endoplasmic reticulum (ER), in both soluble and aggregate forms. Following translation and entry into the ER, the soluble nascent Z-AAT protein is supported by the ER quality control system. Subsequently, the ER clearance pathway–ER-associated degradation (ERAD) –targets the mutant to the ubiquitin-proteasome system (UPS) for degradation.6 Several studies suggest insufficient clearance of Z-AAT results in further retention followed by Z aggregation.7,8 These aggregates are packed into hepatocyte inclusion bodies (IB) (Figure 1). These structures are the histopathological hallmark of AATD liver disease on biopsy and stain positively in hepatocytes with periodic acid–Schiff (PAS) after treatment with diastase (Figure 1). The mechanism of IB formation and their heterogeneity in the distribution and size within hepatocytes (Figure 1) are not clearly understood and elucidated.
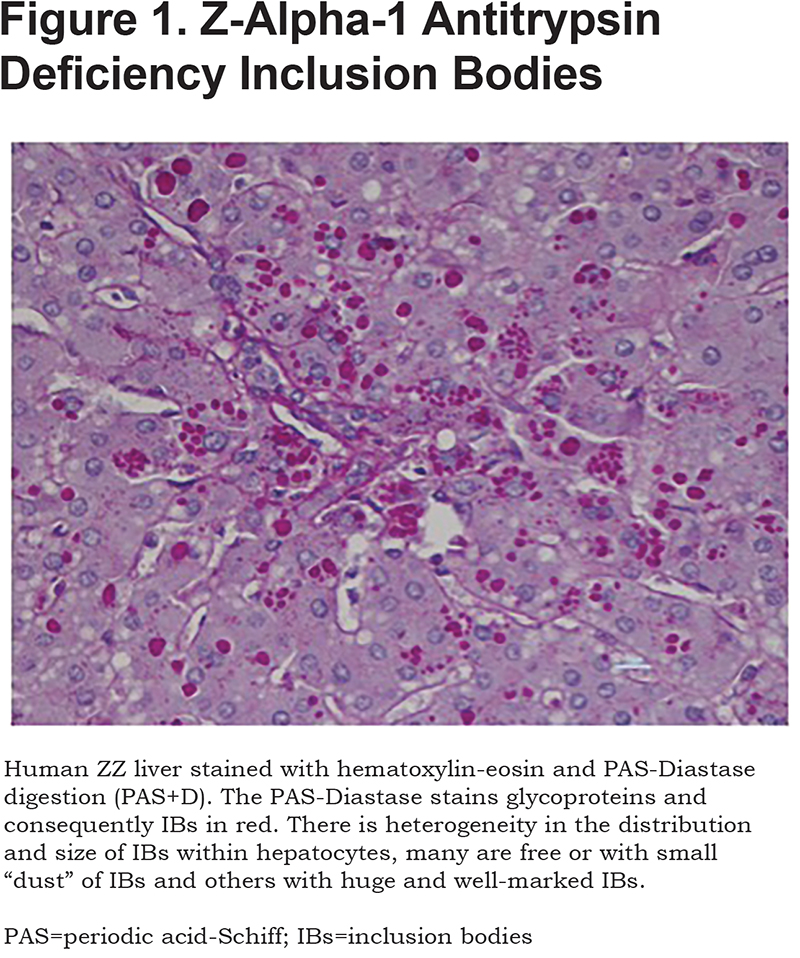
The retention and accumulation of the Z aggregates in the ER present a gain-of-function toxic effect leading to hepatocyte toxicity and initiating events in the pathophysiology of liver disease.9 Based on this idea, preventing production and/or accumulation of Z-AAT was shown to reverse liver injury in disease models.10,11 The concept of Z-AAT gain of toxic function has been supported by the recent findings of Clark et al.12 The authors have evaluated the relationship between positive PAS hepatocytes cells (PAS+D) and fibrosis; they have shown that accumulation of PAS+D increases with progression of the stage of fibrosis. Consequently, only patients carrying AAT variants able to aggregate are susceptible to liver disease and protein retention without aggregation does not lead to liver disease.
Z aggregates, of liver origin, are also present in the serum of all ZZ patients. Consequently, an association between circulating Z-aggregate concentration and liver fibrosis/liver disease has been suggested.13,14 Nevertheless, this association requires further studies to establish whether this biomarker could be useful as a prognostic factor in disease outcomes.
The Z-variant is not the only AAT mutant involved in liver diseases. Rare variants, such as the Mmalton, (Phe51/52 del) and Siiyama (Ser53Phe) variants, may also cause liver disease.15,16 Similar to the Z mutant, these rare variants are able to form aggregates and IBs in the hepatocytes. However, some data have suggested that the process and/or the nature of aggregates involved in these mutants, Z, Mmalton and Siiyama variants, are different.17,18
There is no specific treatment for AATD-associated liver disease, only the standard liver supportive care, such as ursodeoxycholic acid, is used in pediatric practice.19 In general, AATD patients should receive hepatitis vaccinations to prevent further liver injury, follow nutritional management and recommendations and avoid alcohol intake to prevent fatty liver disease and/or obesity.20 However, in severe AATD-related liver disease (liver failure, decompensated cirrhosis), when the disease becomes life-threatening, a liver transplant is performed with excellent survival rates: 90% at 1 year and 80% at 5 years.21
Although the Z protein accumulation is the primary cause of liver damage, this event is not sufficient. Indeed, not all homozygous ZZ patients develop liver injury despite the presence of IBs in the liver.22,23 Therefore, there is likely a significant role for second hits such as genetic and/or environmental modifiers,12,24,25 which determine whether an AATD patient will or will not develop liver injury.
The natural history of AATD is not well defined and liver disease is a lifelong condition.26,27 However, AATD-related liver disease has been described as being different at different stages of life. In childhood, AATD is the most common genetic cause of pediatric liver disease and the most frequent inherited indication for liver transplantation in the pediatric population.28 Over the past 20 years, there has been an increase in the prevalence of the adult form of AATD-mediated liver disease. AATD-related liver disease displays a biphasic pattern with the first peak in an early childhood (birth to 5 years) and the second peak, in adulthood between 50-65 years of age.26 The adult form of the disease seems to be an age-dependent degenerative disease contrary to the pediatric form of the disease, whose severity seems more associated with genetic factors.26,29 Thus, classic confounding factors encountered in childhood, such as breast feeding or other liver diseases (glycogen storage disease or cystic fibrosis), have not been associated so far with this severity.29
In this review, we pinpoint the main, known co-factors of liver disease in AATD in childhood and adulthood.
Why Do Some Patients Do Poorly During Childhood?
Large pediatric cohorts have been investigated for factors responsible for liver disease and have shown that the natural history of AATD is highly variable, ranging from mild elevations in liver blood tests to liver failure necessitating liver transplantation.23,29-31
The main clinical characteristic is usually a prolonged cholestatic jaundice during the first 2 months of life after birth. In an unbiased Swedish study, the authors screened 200,000 newborns in Sweden and detected 127 homozygous ZZ patients in the 1970s and concluded then that only 10% of homozygous ZZ patients develop clinically significant liver disease in the first 4 decades of life and 5% of them have the risk of life-threatening liver disease31,32 (Figure 2). In addition to this pioneer and massive population-based study, more recently the French longitudinal study named DEFI-ALPHA that included only pediatric patients with known AATD irrespective of genotype, observed 18.3% of pediatric AATD patients had a severe liver disease (portal hypertension, presence of oesophageal varices, liver failure, liver transplantation or liver related death) and almost half of them ended up with a liver transplantation.29 As supported by other works, the authors have observed that neonatal cholestasis at diagnosis was significantly associated with severe liver disease. Other signs have also been correlated with worse prognosis such as persistence of elevated liver enzyme, hepatomegaly, and early development of splenomegaly.30,33

Although a small proportion of infants will develop severe liver injury, the majority of individuals will have their liver-related parameters frequently improve; 80% of cholestatic AATD infants will have resolution of the cholestasis and develop normally without clinical evidence of chronic liver disease or presence of advanced liver disease.23,28,34
From the French study, we also learned that liver disease is not restricted to ZZ genotypes; individuals with other genotypes, such as SZ and MMalton variants, may also develop liver damage, especially when associated with other predisposing factors (glycogen storage disease or cystic fibrosis) (Figure 2). Those diseases are already known to be worsened by AATD variants, for instance MZ genotype was shown to aggravate liver disease severity in children with cystic fibrosis.32 However, the inverse is not true, no genetic polymorphisms in CFTR or SERPINA1 genes influence the onset and severity of liver disease in AATD during childhood.35
It is still unknown why patients with the same genotype, even siblings, have such varied clinical courses. It is now well recognized that environmental and/or genetic modifiers, yet to be fully elucidated, are involved in these clinical features. To date, 2 genetic studies, a candidate gene-sequencing strategy24 and a genome-wide association study25 have highlighted some potential genetic modifiers and pinpointed the ERAD pathway as an important player in AATD-related liver disease. Indeed, in 2009, Pan et al demonstrated that difference in ER mannosidase I (ERmanI) expression, a central component of ER quality control, ERAD and consequently, in AAT degradation, was associated with an earlier age-of-onset for end-stage liver disease.24 Recently, our group identified that HERPUD1 R50H and HFE H63D variants are associated with the advanced liver disease component of AATD.25 We also observed that specific pathways, including ERAD and unfolded protein response (UPR), may be risk factors for AATD-caused liver disease.25
However, many other factors are likely to be able to modify AATD-associated liver disease and must be elucidated. Several publications suggest that genetic factors affecting the efficiency of the ERAD/UPS and/or autophagy might act as potential modifiers of AATD- liver disease. There is evidence suggesting that Z-AAT degradation is significantly slower in cells from AATD patients with liver disease than in cells from ATD patients without liver disease.36-38 This suggests that disposal pathways (ERAD/UPS, autophagy) seem relatively inefficient in ZZ individuals presenting liver disease phenotype. Further genome-wide association studies may allow us to explore and resolve the role of these pathways and more importantly identify those genetic modifiers in the future.
Why Do Some Patients Do Poorly During Adulthood?
Liver disease in adults is much more frequent than previously thought. Following a Swedish post-mortem study published by Eriksson, 5%–10% of the ZZ individuals over 50 years of age had developed cirrhosis.9 Similarly, long-term natural history studies of liver disease in AATD have recently shown the prevalence of significant liver fibrosis (F ≥ 2) in ZZ adults is about 30%12,39 and the prevalence of advanced liver disease (cirrhosis, hepatocellular carcinoma) is 10% (Figure 2).40 From 1991 to 2012, 77.2% of transplants for AATD were in adults, and this increased to 87.4% in the last 10 years, suggesting that liver disease is more common in adults than in children. Nevertheless, liver disease in adults is still under-recognized and undiagnosed because there are no specific biochemical and histopathologic markers and analyses to state about AATD-liver disease. All AATD adults should be tested and screened for liver disease given that one-third of adults with ZZ genotype have significant fibrosis without clinical liver disease12 (Figure 2). It is now clear that in ZZ adult males, elevated liver enzymes (upper limit of normal), metabolic syndromes (diabetes) and increasing body mass index (BMI) were identified as risk factors for the development of end-stage liver disease requiring transplantation (Figure 2). 12,27,40,41 The association between obesity and AATD was recently confirmed at the cellular level by comparing AATD epigenetic changes to those linked to liver disease driven by other etiologic exposures (alcohol use, viral hepatitis). AATD and obesity-driven liver disease have marked overlap.42
In contrast, the other classic liver disease co-factors, such as viral hepatitis, autoimmune hepatitis, iron overload, alcohol use or cholestatic liver diseases, and their association with AATD liver disease are still debated. All these co-factors have not been confirmed in all studies.12,27,40,41 Nevertheless, the Z allele may act as a risk factor for cirrhosis in the pathogenesis process of alcoholic and non-alcoholic fatty liver diseases.43,44 Recently, it has also been shown that ZZ carriers had lower serum concentrations of triglyceride, and low and very low-density lipoprotein cholesterol than controls. These results suggest that Z-AATD patients have impaired hepatic secretion of lipids.39 Finally, no statistically significant relationship between the presence of liver fibrosis and severity of COPD was found.12,27,40,41
Interestingly, individuals with liver disease in the neonatal period and who survived childhood do not suffer from liver disease in adulthood. Consequently, liver disease in childhood seems not to directly influence the risk of liver disease in adults.40,45
As observed for the pediatric forms, specific rare AATD (Mmalton) and ZZ and heterozygous Z types are associated with liver fibrosis development, suggesting that the Z variant is a clear and strong disease modifier. Hence, it has been suggested that heterozygosis increases the risk of developing liver disease. The incidence of liver disease could be higher in heterozygotes with the deficiency than in the general population, especially if the affected individuals have other liver comorbidities.5 Many patients who undergo liver transplantation with a diagnosis of AATD are actually heterozygotes who also have other risk factors (alcohol consumption, steatosis).26,46
Chronic liver disease is a major risk factor for the development of liver cancer. The 2 most common forms of primary liver cancer are hepatocellular carcinoma (HCC) and cholangiocarcinoma (bile duct cancer). AATD patients are more likely to develop HCC than cholangiocarcinoma (AATD-mediated cholangiocarcinoma has been reported very rarely). Compared to childhood, in which liver cancer (especially HCC) is reported as extremely rarely, in adults, a post mortem examination has shown that 28% of ZZ individuals had HCC.9 Recently, a longitudinal follow-up of liver function and occurrence of liver disease in 1595 ZZ individuals has found HCC in 2% (29) of the individuals. The overall incidence is 1.3% and this proportion is similar for other liver etiologies such as alcohol.47 Moreover, more than 90% of cases of HCC occur in patients with cirrhosis. Concerning AATD-mediated HCC, in the 29 patients with HCC, the majority (16 patients) had HCC as a primary diagnosis.40 Consequently, ZZ individuals with liver disease are prone to develop HCC and it can occur without preceding cirrhosis.40,48
Why Do Some Patients Do Poorly from a Cellular Pathophysiology Perspective?
Although all the molecular mechanisms underlying hepatocyte damage are not fully understood, we already know that the accumulation of Z aggregates will trigger a cascade of stress pathways that will lead to the death of the hepatocyte and liver injury (chronic hepatitis, cirrhosis, and HCC) (Figure 3). Indeed, the apoptosis pathway is induced potentially through mitochondrial dysfunction.49 The induction of this death pathway is closely associated with the accumulation of the Z variant and has an important impact on the liver disease process. Hepatocytes expressing the Z aggregates have an increased caspase-activated mitochondrial depolarization and are more prone to apoptosis (Figure 3).50 Therefore, the hepatocytes containing Z IBs are impaired in cell proliferation compared to the cells without or with lesser Z accumulation/IB which are promoted to proliferate to preserve the liver cell mass. In a vicious cycle of death and regeneration, the hepatic stellate cells are activated which initiates the hepatic fibrosis process. This hypothesis was also supported by experiments in which normal hepatocytes were found to have a selective proliferative advantage in the liver when transplanted into a AATD mouse model.51
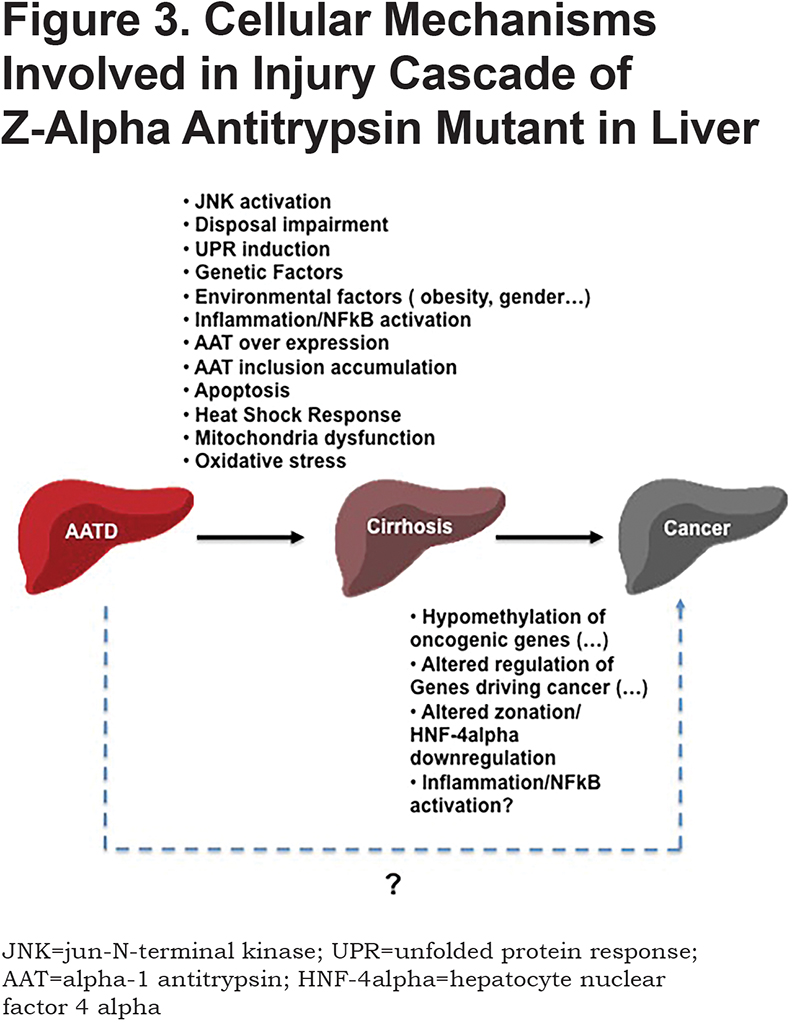
Activation of NFκB is another hallmark of the cellular response to Z accumulation (Figure 3). Little is known about the mechanism underlying the activation of this pathway but it was suggested that ER morphological changes caused by the accumulation of Z aggregates might disrupt ER calcium homeostasis leading to release into the cytosol and NFκB activation.52-54 In a mouse model, it was also observed that NFκB activation could prevent fibrosis associated with Z-AATD. Z accumulation and hepatic fibrosis were worse in the Z-AATD mouse model crossed with 2 different mice models deficient in NFκB signaling.11,55 The regulation of NFκB signaling on autophagy might explain its protective role in the liver fibrosis.
In contrast to the NFκB pathway activation, Z mutation and/or aggregations fail to induce the UPR, a stress response that controls ER homeostasis through transcriptional activation of several genes involved in ER folding, degradation and export.54 Nevertheless, Z accumulation seems to sensitize the cell to second insults that can cause a much stronger UPR response than observed into WT cells(Figure 3).25,54
Curiously, another well-known pathway, induced following protein aggregation– the heat shock response (HSR) pathway– is activated in cells expressing the Z mutant. The down-regulation of this stress response, through silencing of HSF1, the master regulator of the HSR, is increased maturation and secretion of the mutant Z-AAT.56 The mechanism by which the silencing of HSF1 could be beneficial for AATD still needs to be elucidated but these results suggest a link, a cross talk between the ER compartment and the cytoplasmic stress management by HSR.56
Other cellular pathways such as the oxidative stress or c-Jun N-terminal kinase (JNK) and c-Jun signaling (Figure 3) are also induced in response to Z accumulation.57,58 The oxidative stress pathway has been shown to be a contributing factor in the development of liver damage in animal models of AATD. Higher levels of reactive oxygen species and a more oxidized, cellular redox state were observed in liver tissue from AATD mice as compared to WT mice.58 JNK signaling was also found to be involved in the development of liver damage. JNK signaling, through the transcription factor c-Jun, is involved in transcriptional regulation of SERPINA1 and inhibition of JNK was found to reduce Z accumulation.57
Environmental factors modulating SERPINA1 expression may also potentially affect liver injury. AAT is an acute-phase protein that overexpresses (by 2- to 4-folds) in response to inflammatory induction.59 This overexpression could consequently increase the amount of Z aggregate forms and increase the burden of Z-AAT hepatotoxicity. In agreement, treatment with indomethacin, a nonsteroidal anti-inflammatory drug, in an AATD mouse model was found to up-regulate AAT expression and enhance liver injury.60
Finally, the pathogenesis involved in liver cancer development is still unclear and needs to be elucidated (Figure 3). Nevertheless, some events described by different groups might play a role in carcinogenesis (Figure 3). In an AATD mouse model, a down-regulation of hepatocyte nuclear factor 4 alpha (HNF-4α) has been observed to be associated with a loss of zonation.61 Given that HNF-4α is a master transcription factor in hepatocyte phenotype maintenance62 and has a suppressor role in liver cancer,63 its down-regulation could favor HCC development. Others have also observed a significant genomic hypomethylation in AATD liver-impacting genes related to liver cancer following a DNA methylation study on liver biopsies of 118 ZZ-AATD adult patients (104 without cirrhosis, 14 cirrhotic).42 This up-regulation of markers related to liver cancer was also described by Segeritz et al. Based on a comparative "omics" approach, the authors have observed an up-regulation of specific proteins associated with predisposition to malignancy in human ZZ-induced pluripotent stem cells.64 Finally, NFκB activation-mediated liver inflammation might also play a role in carcinogenesis.65
Conclusion
AATD-mediated liver damage has been linked to a proteotoxicity resulting in the accumulation of misfolded AAT in the ER of hepatocytes triggering a cascade of stress responses and eventually, liver cell death. There is a high degree of clinical variability in the disease outcome and so far, we have not been able to screen individuals at risk of developing liver damage. Nevertheless, recent long-term natural history studies of liver disease in AATD have given insights into AATD-mediated liver damage and some clues to answer the questions raised in the manuscript's title: why do some patients do poorly? what do we know so far?
AATD-related liver disease displays a biphasic pattern with the first peak in early childhood (birth to 5 years) and the second peak, in adulthood between 50-65 years of age.26
Beginning in childhood, only 10% of homozygous ZZ patients develop clinically significant liver disease in the first 4 decades of life.31,32 Neonatal cholestasis at diagnosis is significantly associated with severe liver disease (Figure 2). Liver disease is not restricted to ZZ genotypes; individuals with other genotypes such as SZ and Mmalton variants may also develop liver damage (Figure 2). Even so, it is still unknown why patients with the same genotype, even siblings, have such varied liver clinical courses. It is now well recognized that environmental and/or genetic modifiers, yet to be fully elucidated, are involved in these clinical features. To date, 2 genetic studies24,25 have highlighted some potential genetic modifiers and pinpointed the ERAD pathway as an important player in AATD-related liver disease (Figure 3).
In adulthood, AATD-liver disease is amuch more frequent occurrence than previously appreciated. It is now clear that in ZZ adult males over 50 years of age, elevated liver enzymes (upper limit of normal), metabolic syndromes (diabetes) and increasing BMIs are identified as risk factors for the development of end-stage liver disease requiring transplantation (Figure 2).12,27,40,41 Finally, as observed for the pediatric forms, rare AATD genotypes such as Mmalton and heterozygous Z are associated with liver fibrosis development (Figure 2).
Consequently, given all this information, for any child with neonatal cholestasis, a diagnosis of AATD must be considered. Similarly, adult males over 50 with obesity and metabolic syndromes should be tested. The observed commonalities between liver disease and obesity, for instance, suggests that all the patricians should advise patients to lose weight, eat healthy, and avoid alcohol intake and recommend immunization against hepatitis in order to prevent the development of end-stage liver disease in later adulthood.
Clearly these findings have increased our understanding of the disease, nevertheless, many challenges still need to be addressed and consequently, further research is required to identify potential disease-genetic modifiers, unknown factors that lead to increased susceptibility to HCC and molecular mechanisms involved in the disease pathogenesis. These challenges need to be resolved for the development of early and non-invasive diagnosis, and to develop new therapeutic strategies. Indeed, in contrast to the lung disease, for which an augmentation therapy is available for treating frequent and severe pulmonary exacerbations, there are currently no therapies for AATD-associated liver disease beyond transplantation.66 Nevertheless, autophagy enhancers, small molecule chaperones, gene repair (CrisprCas9) associated with cell transplantation and siRNA technologies hold promise for the future treatment of this disease.66
Acknowledgements
Grant support was provided by the Association Nationale Francaise Deficit en Alpha-1 Antitrysine , Alpha-1 Association Suisse CSL Behring, the University of Bordeaux, the National Institute of Health and Medical Research (INSERM, and the National Center for Scientific Research (CNRS).
Declaration of Interest
MB declares no conflicts of interest.