Running Head: Design of the RETHINC Study
Funding Support: Support for this study includes a National Heart, Lung and Blood Institute grant HL128952 and HL128954. The study drug was supplied by Novartis Pharmaceuticals. Additional financial support to conduct the trial was provided by contributions made through the COPD Foundation from Sunovion Pharmaceuticals.
Date of Acceptance: June 16, 2020
Abbreviations: Redefining Therapy in Early COPD, RETHINC; COPD Assessment Test, CAT; forced expiratory volume in 1 second, FEV1; forced vital capacity, FVC; SubPopulations and InteRmediate Outcome Measures In COPD Study, SPIROMICS; St George’s Respiratory Questionnaire, SGRQ; Baseline Dyspnea Index, BDI; Transition Dyspnea Index, TDI; area under the curve, AUC; forced expiratory flow, FEF; chronic obstructive pulmonary disease, COPD; Global initiative for chronic Obstructive Lung Disease, GOLD; Canadian Cohort of Obstructive Lung Disease, CanCOLD; COPD Genetic Epidemiology study, COPDGene®; computed tomography, CT; modified Medical Research Council, mMRC; long-acting beta2-agonist, LAMA; long-acting muscarinic antagonist, LAMA; Food and Drug Administration, FDA; slow vital capacity, SVC; inspiratory capacity, IC; National Heart, Lung and Blood Institute, NHLBI; Pulmonary Trials Consortium, PTC
Citation: Han MK, Ye W, Kim DY, Woodruff P; Pulmonary Trials Cooperative investigators. Design of the redefining therapy in early COPD study. Chronic Obstr Pulm Dis. 2020; 7(4): 382-389. doi: http://doi.org/10.15326/jcopdf.7.4.2020.0157
Online Supplemental Material: Read Online Supplemental Material (251KB)
Introduction and Rationale
Chronic obstructive pulmonary disease (COPD) is typically defined based on a reduced ability to forcefully exhale. Hence, airflow obstruction, measured by spirometric assessment via the ratio of the forced expiratory volume in 1 second (FEV1) to the forced vital capacity (FVC) after bronchodilator is inherent to the diagnosis of COPD.1 However, we have previously reported that some smokers without airflow obstruction report increased respiratory symptoms similar to individuals who have airflow obstruction. In the SubPopulations and InteRmediate Outcome Measures In COPD Study (SPIROMICS) cohort,2 current or former smokers with respiratory symptoms as defined by COPD Assessment Test (CAT) score ≥ 10, experienced significantly higher rates of “COPD-like” exacerbations both as compared to non-obstructed individuals with lower CAT scores as well as Global initiative for chronic Obstructive Lung Disease (GOLD)1 stage 1-2 participants with low CAT scores. Follow-up SPIROMICS data analysis demonstrated increased total mucin concentrations among the symptomatic non-obstructed smokers versus asymptomatic individuals.2 In the Canadian Cohort of Obstructive Lung Disease (CanCOLD) cohort, exacerbation events in unobstructed participants resulted in an increased rate of missed social activities, missed work for income and inability to do housework suggesting that such events have real clinical significance.3 Similarly, in the COPD Genetic Epidemiology (COPDGene®) cohort, respiratory-related impairments were found in 54% of the non-obstructed current and former smokers including greater St George’s Respiratory Questionnaire (SGRQ) scores, lower 6-minute walk distances and computed tomography (CT) evidence of emphysema or airway wall thickening.4
Notably, among these symptomatic current or former smokers in the SPIROMICS cohort, 42% had been prescribed bronchodilators and 23% inhaled glucocorticoids. However, currently there is no clinical trial evidence to guide treatment in such patients. This provides the rationale for the RETHINC study where the following overarching hypothesis is being tested: current and former smokers with respiratory symptoms despite normal spirometry (FEV1/FVC ≥ 0.70) will derive benefit from inhaled bronchodilator therapy, even though they are excluded from the current treatment guideline recommendations. The GOLD consensus report1 and COPD Foundation guidelines5 currently recommend the use of long-acting bronchodilators, used either alone or in combination, as treatment for patients with COPD with any severity of lung dysfunction who have symptoms based on CAT ≥ 10 or modified Medical Research Council dyspnea score (mMRC) ≥ 2. Long-acting bronchodilators include the long-acting beta2-agonists (LABAs) as well as several long-acting muscarinic antagonists (LAMAs) that are currently Food and Drug Administration (FDA)-approved in the United States as monotherapy for the treatment of COPD. The FDA has also approved the use of dual bronchodilators which combine a LABA and a LAMA for maintenance therapy in COPD.
In RETHINC, our study drug is the dual bronchodilator indacaterol/glycopyrrolate. Indacaterol/glycopyrrolate has rapid onset of action with sustained bronchodilator effect over a 12-hour period and is administered twice daily. It is indicated for long-term maintenance treatment of COPD including chronic bronchitis and/or emphysema. In clinical studies, both LABAs and LAMAs appear to be effective for improving lung function in terms of FEV1 although the effects of a combination LABA and LAMA are greater than the effects of either drug alone. In the FLIGHT 1 and FLIGHT 2 studies comparing indacaterol/glycopyrrolate 27.5/15.6 mcg twice daily to indacaterol 27.5 mcg twice daily, glycopyrrolate 15.6 mcg twice daily or placebo, the improvement in FEV1 area under the curve from 0 to 12 hours (AUC0-12h) for the combination was superior to either monocomponent (p < 0.001).6 Symptom improvement based on SGRQ and Transition Dyspnea Index (TDI) scores as well as rescue medication use were also significantly greater in the combination treatment group as compared both to monocomponents and placebo (p < 0.001). Another advantage of dual bronchodilators in COPD is that individuals may respond preferentially to 1 of the 2 classes of bronchodilators. Hence, in choosing a bronchodilator to test in this study, we chose a bronchodilator therapy with dual mechanism of action which is most likely to produce the greatest bronchodilation and symptom improvement in the most number of individuals, allowing us to best test the hypothesis that bronchodilation in symptomatic smokers will result in respiratory symptom improvement. By using maximal bronchodilator therapy, we have the best chance of proving or disproving our hypothesis.
Methods
Study Design
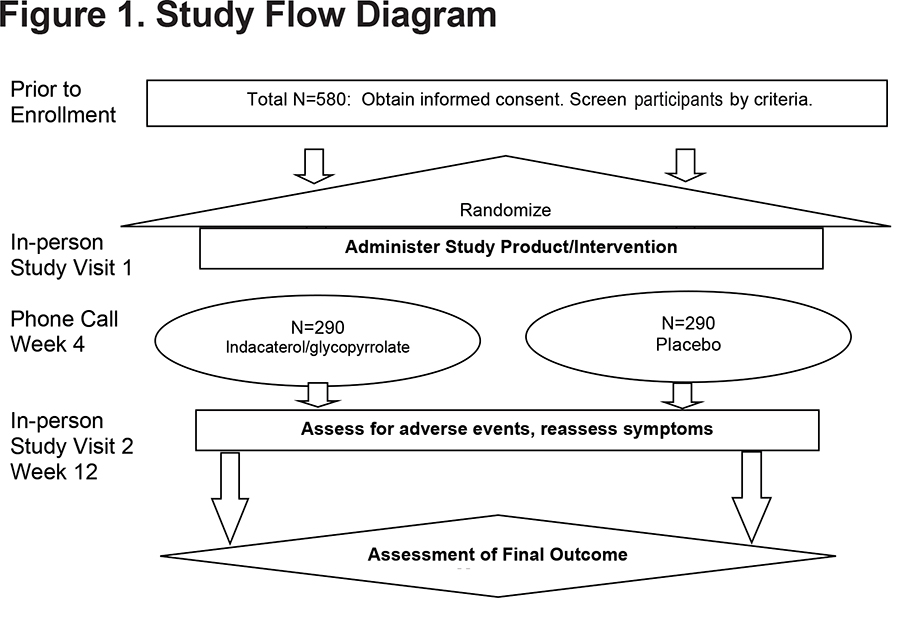
RETHINC is a multi-center Phase 3, double-blind, placebo-controlled, parallel-arm study comparing indacaterol/glycopyrrolate 27.5/15.6 mcg inhaled twice daily versus placebo in participant with a ≥ 10 pack-years smoking history with post-bronchodilator FEV1/FVC ≥ 0.70 and CAT ≥ 10. Treatment duration is 12 weeks with 290 patients in each arm. SGRQ, CAT, Baseline Dyspnea Index (BDI) and TDI, and spirometry will be performed at baseline and 12 weeks. The BDI measures the severity of dyspnea at the beginning of the clinical trial. The TDI measures changes from this baseline (transition period) at subsequent visits. A phone call at 4 weeks will assess for adverse events. Participating centers included a mix of academic, Veterans Affairs and community medical centers. Institutional review board approval was obtained at every participating center. Informed consent was required of every participant.
The primary outcome will be based on an intention-to-treat population for a composite endpoint. The composite endpoint will be comparison of the proportion of individuals on treatment versus placebo who experience a 4-unit change in SGRQ after 12 weeks without treatment failure during the 12-week study period. Treatment failure is defined as an increase in lower respiratory symptoms necessitating treatment with a long-acting bronchodilator, corticosteroids or antibiotics. Treatment failure will be determined by a blinded assessment. If we meet our primary outcome, we will conclude that treatment improves clinical status in symptomatic “at risk” smokers even if they do not meet the formal spirometric definition of COPD (FEV1/FVC < 0.70). Important secondary endpoints in comparing treatment versus placebo after 12 weeks will be proportion of individuals with 2-unit change in CAT without treatment failure; proportion of individuals with a 1-unit change in BDI/TDI without treatment failure; proportion of individuals with a 4-unit improvement in SGRQ and a 1-unit improvement in BDI/TDI without treatment failure; mean change in SGRQ; mean change in CAT; AUC0-3h in FEV1 after study drug; change from baseline in 12-hour trough FEV1; change from baseline in 12-hour trough inspiratory capacity, FEF25-75 and iso-volume FEF25-75 and mean change symptoms and rescue medication use based on daily diary; and treatment failure defined by increase in lower respiratory symptoms necessitating treatment with active, long-acting inhaled bronchodilator, corticosteroids or antibiotics.
A permuted block randomization scheme will be used with varying block sizes stratified by clinical center, smoking history (current smoker/former smoker) and prior maintenance treatment for COPD requiring washout. Participants will be randomized to receive active drug and placebo with equal probability (1:1), via the internet with a central interactive response system.
Inclusion and Exclusion Criteria
The inclusion and exclusion criteria are provided in the Appendix 1 in the online supplement as well as a Table of Prohibited Medications in Appendix 2 in the online supplement.
Baseline and Follow-up Assessments
For patients meeting inclusion and exclusion criteria (Appendix Table 1 in the online supplement) who are not on any prohibited medications (Appendix Table 2 in the online supplement) or prohibited COPD medications (Appendix Table 3 in the online supplement), the visit schedule is outlined in Table 1. Participants taking any prohibited COPD medication (Appendix Table 3 in the online supplement) are required to undergo medication washout prior to enrollment. For these participants who require wash-out, see Appendix Table 4 in the online supplement for a schedule of events including an additional on-site washout visit. Participants who tolerate wash-out may proceed to enrollment. Once randomized, participants will undergo study-related procedures as outlined in Table 1. The Baseline Visit consists of questionnaires and spirometry. The spirometry performed at enrollment includes a pre-and post-bronchodilator slow and forced vital capacity maneuver. This post-bronchodilator spirometry is performed after administering 4 puffs of albuterol. Questionnaires include the CAT and SGRQ which are self-administered using paper. However, this study is implementing a self-administered electronic version of the BDI/TDI to maximize the robustness of the data.7,8 A paper symptom diary is also distributed to the participant at this visit. Overall study visit flow is summarized in Figure 1.
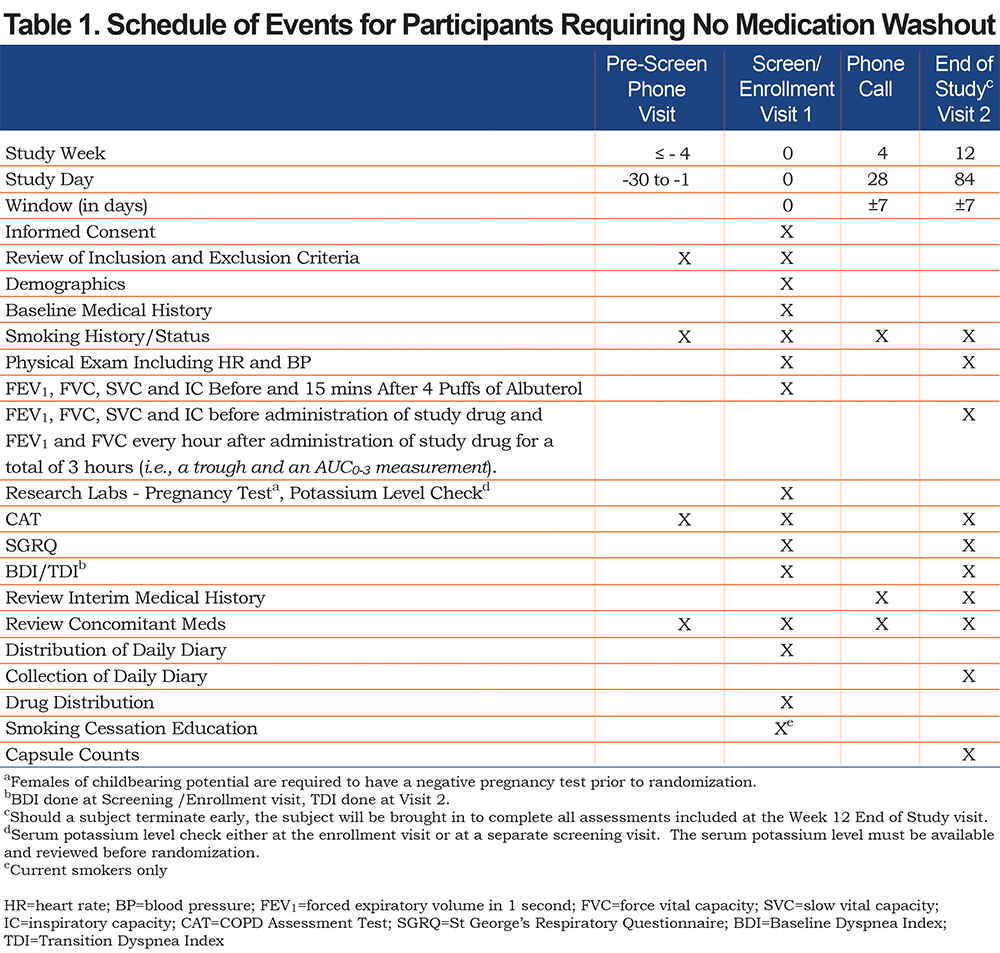
All participants undergo a phone call at 4 weeks to review smoking status, interim medical history and assess for adverse events. Adverse events are defined as any untoward or unfavorable medical occurrence associated with an individual’s participation in the research. Adverse events reported in the package insert approved by the FDA for indacaterol/glycopyrrolate occurring at an incidence greater than or equal to 1% higher than placebo included nasopharyngitis, hypertension, back pain and oropharyngeal pain. At 12 weeks, participants repeat the CAT, SGRQ and BDI/TDI. At this visit, forced and slow vital capacity maneuvers are performed before administration of study drug and then FEV1 and FVC are performed every hour after administration of study drug for a total of 3 hours (i.e., a trough and an AUC0-3 measurement).
Statistical Analysis and Sample Size Considerations
The primary outcome will be based on the intention-to-treat population for the SGRQ measurement which will be supplemented by blinded assessment of treatment failure in a composite endpoint. For individuals who do not complete 12 weeks of treatment, we will make every effort to bring them back at 12 weeks for SGRQ and other symptom and physiologic assessments. For those who drop out of the study and decline any further visits, we will compare baseline values to those who complete the 12-week assessments and note any differences that could impact the analysis. Our composite primary endpoint definition will allow us to minimize a major source of missing data due to “drop-outs” related to disease worsening and subsequent lack of data on SGRQ score at 12 weeks. If those drop outs occurred because of treatment failure (which may be disease related and intervention related), then they will still be informative in that they add to our primary outcome because they will meet criteria for “treatment failure.” There will remain some drop-outs that are non-informative because they are due to factors other than treatment failure.
The primary analysis will be conducted via Generalized Estimating Equestions regression with logit link to compare the proportion of participants who experience improvement with treatment defined as a 4-point drop in SGRQ and absence of treatment failure in the treatment versus placebo groups, adjusted for clinical center of recruitment, change of smoking status between baseline and 12 weeks, prior maintenance treatment for COPD requiring washout, and body mass index at baseline. No formal statistical analysis for efficacy will be conducted until the end of the study given the interest in obtaining follow-up for multiple endpoints of interest.
We performed sample size estimations using preliminary data from the Novartis FDA development program studies. At week 12, using FLIGHT1 where the effect on SGRQ was slightly smaller than FLIGHT 2, 57% of participants taking indacaterol/glycopyrrolate versus 39% of participants taking placebo had a ≥ 4 unit decrease in SGRQ.6 Using these data, we estimate (with a 2-sample test of proportions) that 290 participants per arm would provide 90% power with the following assumptions: (1) a type I error rate of .05; (2) 10% loss to follow-up and (3) approximately 20% lower effect size given the FLIGHT studies were based on a more severe patient population. We do not have data available on the effect size of bronchodilators in the GOLD 0 population or the specific effect that our composite outcome (including the treatment failure definition) will have. However, we believe that a sample size estimate using this approach should provide a realistic estimate of power given that we are assuming a lower effect size and protecting against bias towards the null result.
Study Organization and Funding
Along with the participating clinical centers, RETHINC has several governing bodies as it is being performed via the National Heart, Lung and Blood Institute (NHLBI)-funded Pulmonary Trials Consortium (PTC) framework. In addition to the National Institutes of Health’s NHLBI Project Office, the PTC framework for this study also includes a Data Coordinating Center at the University of Michigan and the Network Management Core at the University of Pittsburgh. There is also a Data Safety Monitoring Board to provide annual and ad hoc evaluations of the study with recommendations to the NHLBI and investigative team. More information about the PTC can be found here https://www.pulmonarytrials.org/about-pulmonary-trials-cooperative. Support for this study includes an NHLBI grant HL128952 and HL128954. Study drug was supplied by Novartis Pharmaceuticals. Additional financial support to conduct the trial was also provided by Sunovion Pharmaceuticals.
Discussion
Prior to 2006, the GOLD consensus statement included a “GOLD 0” stage that was defined as symptomatic, at-risk individuals who did not have airflow obstruction.9 However, in 2007, the GOLD committee removed GOLD 0. This decision was informed in part by data from the population-based Copenhagen City Heart Study where many individuals without symptoms still progressed to airflow obstruction.10,11 Roughly 13% and 21% of symptomatic, non-obstructed smokers in that cohort developed COPD at 5 and 15 years follow-up.12 By contrast, 12% and 19% of asymptomatic, non-obstructed smokers at enrollment developed COPD by 5 and 15 years, respectively. Hence, concern was raised that the GOLD 0 stage may not help in identifying individuals at risk for development of obstruction.
There has been much discussion recently regarding the meaning and definition of early COPD. All of these potential definitions, however, focus on individuals who either have or are likely to develop spirometric obstruction. We would argue, however, that whether or not such individuals go on to develop spirometric airflow obstruction, these symptomatic smokers have clinically significant exacerbations and CT abnormalities that suggest subclinical airway disease. Thus, these individuals have disease that should be recognized as such and which may warrant treatment.
Given that spirometry is vastly underutilized in primary care, it is not surprising that a large percentage of smokers with symptoms despite preserved spirometry are already being treated with COPD-specific respiratory medications. However, we currently have no evidence basis upon which to guide such decisions. The RETHINC study is the first step to understanding whether therapies approved for COPD benefit those without traditionally defined airflow obstruction. We have chosen to study a dual bronchodilator not because we ultimately believe that this necessarily would be first line therapy for such patients, but rather to maximize our ability to determine whether this is a bronchodilator responsive disease.
We admit to several limitations to this study. First, an improvement in symptoms over 12 weeks does not guarantee sustained responses over the longer term. However, the studies used for FDA registration of the study drug were performed over 12 weeks and this relatively short trial duration enhances the comparability and feasibility of the RETHINC study. The second is that it is possible that the clinically meaningful thresholds we have predetermined for SGRQ, CAT and BDI/TDI that were defined in patients with COPD may not be the correct thresholds for individuals with milder disease. We also note that there is the potential that bronchodilator therapy may reduce exacerbations but not influence shorter term outcomes assessed at 12 weeks, which could bias our study towards the null hypothesis.
The RETHINC trial will be the largest study of its kind to address pharmacologic treatment of symptomatic smokers without obstruction. Data from the CanCOLD population-based study suggests this may represent roughly 25% of smokers. Regardless of the results, this trial will be informative. If the result is positive, then we will have defined at least 1 effective treatment approach. If the result is negative, we will have evidence that the current GOLD treatment pathway should not be generalized to this population and further subgroup analyses conducted to determine whether a subset of this study population might still be candidates for future prospective bronchodilator investigation and in whom additional therapies need to be investigated. Given the substantial gap in knowledge regarding this patient population, RETHINC will also yield a range of novel clinical data through analysis of secondary outcomes which should be useful for planning future studies.
Acknowledgments
The authors thank the RETHINC participants and participating physicians, investigators and staff for making this research possible. The authors wish to acknowledge support of this study from the NHLBI and the Pulmonary Trials Consortium including the Network Management Core and Pulmonary Trials Consortium investigators and staff. Also supporting this study is the Data Safety Monitoring Board including Gordon Bernard, MD; Joao A. De Andrade, MD; Deborah R. Barnbaum, PhD; Daren L. Knoell, PharmD; Peter Lindenauer, MD, MSc; Andre Rogatko, PhD; and Marinella Temprosa, PhD. The authors also wish to acknowledge support from Novartis for supply of the study drug and to Sunovion for financial contributions made through the COPD Foundation.
Declarations of Interest
MKH reports consulting income from GlaxoSmithKline, AstraZeneca, Boehringer-Ingelheim, Merck and Mylan outside the submitted work as well as provision of study drug from Novartis for this study and research support for this study from Sunovion via the COPD Foundation. WY and DK report no conflicts. PW reports personal fees from GlaxoSmithKline, NGM BioPharmaceuticals, Amgen, Glenmark Pharmaceuticals, Theravance, Clarus Ventures, Astra Zeneca, 23andMe, Sanofi, Regeneron and Genentech outside the submitted work.