Running Head: AATD: Characterization of the Mutation Leu353Phe_fsTer24
Funding Support: Yohan Bossé holds a Canada Research Chair in Genomics of Heart and Lung Diseases. François Maltais was the recipient of the CIHR/GSK research Chair on COPD at Université Laval. This study was partly supported by grants from the Chaire de pneumologie de la Fondation JD Bégin de l’Université Laval, the Fondation de l’Institut universitaire de cardiologie et de pneumologie de Québec, and the Canadian Institutes of Health Research (MOP – 123369) and funding from Grifols to the Catalan Center for Research in Alpha-1 antitrypsin deficiency of the Vall d´Hebron Institut de Recerca (VHIR) in the Vall d´Hebron Barcelona Hospital Campus, Barcelona, Spain. The sponsors had no role in study design, data collection, data analysis, data interpretation, or writing of the report.
Date of Acceptance: August 14, 2020 | Published Online Date: November 2, 2020
Abbreviations: alpha-1 antitrypsin deficiency, AATD; alpha-1 antirypsin, AAT; protease inhibitor, PI; base pair, bp; wild type, WT; insertion of nucleotide, insT; computed tomography, CT; allele-specific polymerase chain reaction, AS-PCR; forced expiratory volume in 1 second, FEV1; forced vital capacity, FVC; diffusion capacity to carbon monoxide, DLCO
Citation: Bellemare J, Gaudreault N, Valette K, et al. The clinical utility of determining the allelic background of mutations causing alpha-1 antitrypsin deficiency: the case with the null variant Q0(Mattawa)/Q0(Ourém). Chronic Obstr Pulm Dis. 2021; 8(1): 31-40. doi: http://doi.org/10.15326/jcopdf.8.1.2020.0168
Introduction
Alpha-1 antitrypsin deficiency (AATD) is inherited as an autosomal codominant disorder associated with reduction and/or dysfunction of the circulating protein alpha-1 antitrypsin (AAT). AAT acts as the main serine protease inhibitor protecting the lung parenchyma against proteolytic attack by neutrophils during inflammation.1 AATD is caused by mutations in the SERPINA1 gene. SERPINA1 is a 13.9 kb gene located on chromosome 14q32.13, composed of 3 non-coding exons (IA, IB, IC) and 4 coding exons (II to V). This gene encodes for AAT, consisting of a 394 amino acid (52 kDa) glycoprotein in its mature form. Using nephelometry, AAT is normally expressed at serum levels of 0.9 to 2.0g/L.2-4 Over the years, 0.6g/L or 11μM has been proven to be the minimum plasma protective threshold to protect the lung.2,5-8
Hundreds of genetic variations in the SERPINA1 gene have been described to date with a range of effects, including deficient and null alleles.9 The most common deficient alleles are S (E264V, rs17580) and Z (E342K, rs28929474) that lead, in their homozygote forms, to AAT levels of 40%-50% and 15% of normal value, respectively. Null homozygotes are rare but lead to undetectable circulating AAT and are, therefore, of high clinical relevance.
The investigation algorithms to detect AATD usually consist in a stepwise approach starting with the measurement of serum AAT levels performed by nephelometry or other immuno-based methods. Phenotyping protease inhibitor (PI) with isoelectric focusing of serum AAT is still used in most diagnostic algorithms in combination or not with targeted genotyping of the most common variants.10,11 However, PI phenotyping and genotyping lack resolution and completeness, respectively, and may, therefore, be inconclusive. The gold standard to detect AATD is DNA sequencing of SERPINA1, which provides complete and precise information about the genetic variants including both common and rare variants. The availability of accurate methods to identify AATD such as DNA sequencing is of primary importance in order to provide correct diagnosis and allow adequate follow-up and treatment, particularly when dealing with variants other than E264V (S) or E342K (Z).12-14
The rare variant of interest in this study is Leu353Phe_fsTer24 (rs763023697). This variant was first described by Cox and Levison in 1988 in 3 siblings of a large family in Canada and labelled Q0Mattawa.15 In 1989, it was molecularly characterized in the same family by Curiel et al16who showed that an insertion of 1 nucleotide at position 353 of the protein resulted in a premature stop codon of 24 amino acids downstream (Leu353Phe_fsTer24) on a M1 (Val213) background. The resulting impact is a truncated and unstable protein degraded in the hepatocytes, thereby undetectable in serum.15 This variant was also identified in several families in Europe.9,17,18 In 2002, Seixas et al19 described the null allele Q0Ourém from 2 siblings born in Ourém and having the same T-insertion mutation as Q0Mattawa on an M3, instead of an M1 (Val213) background. There are no clinical implications of distinguishing between the M1 or M3 background. However, for a patient heterozygote for the Leu353Phe_fsTer24 variant, the background (M1 or M3) must be determined by DNA sequencing of each independent allele in order to differentiate whether this person inherited Q0Mattawa or Q0Ourém.
In this article, we report 7 new cases carrying the rare Leu353Phe_fsTer24 null variant, including 2 who also carry E264V (S) or E342K (Z). The objective of this study was to develop an allele-specific DNA sequencing method in order to determine the M1 (Q0Mattawa) or M3 (Q0Ourém) background of the Leu353Phe_fsTer24 variant in our 7 cases and also to elucidate whether E264V (S) and E342K (Z) observed in 2 of these cases were on the same or distinct allele than Leu353Phe_fsTer24. For these 2 patients, the 2 mutations on the same allele (inherited from one of the parents) or on distinct alleles (inherited from both parents) had obvious clinical implications as this implies a diagnostic of heterozygote in the former (mild to intermediate AATD) or compound heterozygote in the later (more severe AATD).
Materials and Methods
Participants and Ethics
The 7 study participants were identified from 2 clinical practices: Quebec City, Quebec, Canada and Barcelona, Spain. Demographic data, smoking history, lung function, and chest computed tomography (CT) data were abstracted from medical reports. All participants provided informed consent and the study protocol was approved by the Research Ethics Committee of the Institut universitaire de cardiologie et de pneumologie de Québec, Quebec City, Canada.
AAT Serum Levels and DNA Sequencing of SERPINA1
Blood samples were drawn by venipuncture (Vacutainer Lavender top EDTA collection tube) and serum aliquots were stored at -80°C within 2 hours of collection. AAT serum levels were measured by immunoturbidimetry on a COBAS INTEGRA 800 analyser (Roche Diagnostics, Laval, Canada). DNA was extracted from 200µL of frozen buffy coat using QIAamp Blood Mini kit, Qiagen, Ontario, Canada. DNA sequences of the coding regions (i.e., 4 exons) of the SERPINA1 gene were obtained by Sanger sequencing as previously described.13
Allele-specific Polymerase Chain Reaction for Leu353Phe_fsTer24 Allelic Background Determination
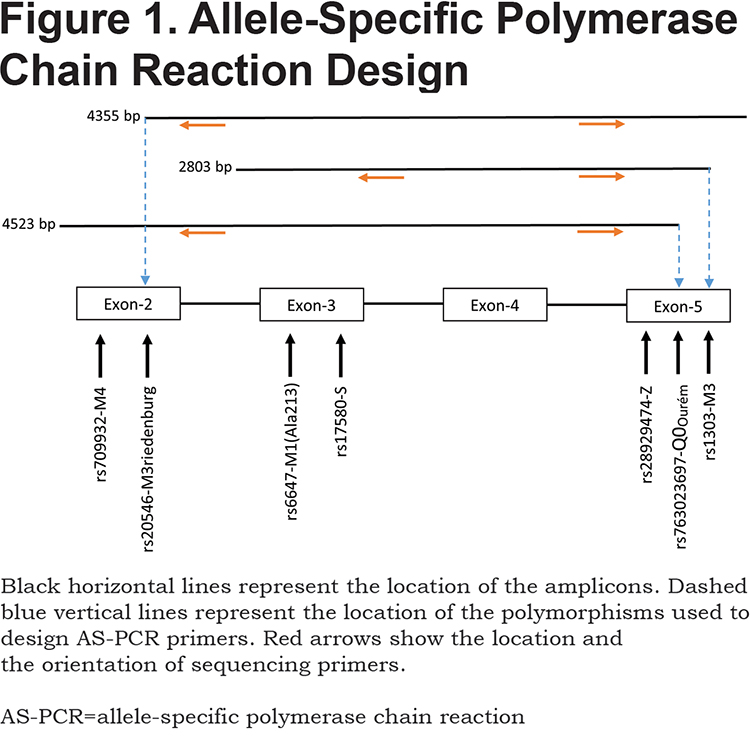
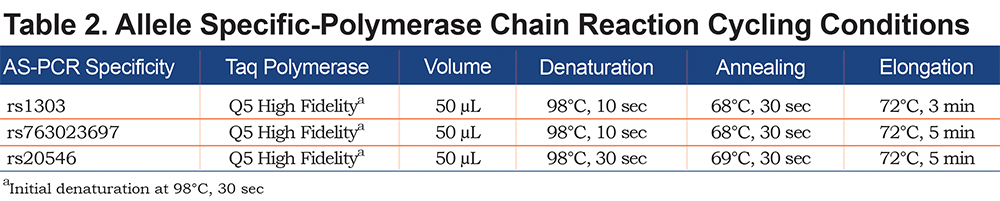
Allele-specific (AS) polymerase chain reaction (PCR) was performed in order to amplify each allele independently. Based on the heterozygosity of a single polymorphism, forward AS-primers and 1 common reverse primer were designed (Figure 1). Polymorphism rs1303 (M3) was used to design the AS-PCR (2803 bp) of cases 1, 2, 3, 6 and 7. Cases 4 and 5 are homozygotes for rs1303 based on the initial Sanger sequencing of the coding regions. It was, thus, not possible to use rs1303 for AS-PCR of these 2 cases. Polymorphisms rs763023697 (Leu353Phe_fsTer24) and rs20546 (M3Riedenburg) were used to design the AS-PCR of cases 5 (4523 bp) and 4 (4355 bp), respectively. Table 1 shows the AS-PCR primers. Primers were purchased from Integrated DNA Technologies, Iowa, United States. Q5 High-Fidelity DNA Polymerase was from New England Biolabs, Ontario, Canada and dNTP mix was from ThermoFisher Scientific, Ontario, Canada. AS-PCR was performed where each allele was specifically amplified in a single, independent tube. Amplification was performed in a volume of 50µL containing 100 ng of genomic DNA, 1X of PCR Buffer and Q5 High GC Enhancer, 200nM of each primer, 200µM of each dNTP and 1 Unit of DNA polymerase. Amplification was carried out in a GeneAmp® PCR System 9700 (Applied Biosystems), using 35 cycles as described in Table 2. The sequencing reaction was then performed on each allele independently, using standard procedures and run on the ABI 3730xl DNA Analyzer (Applied Biosystems). Sequencing primer 5’-GGGAGTGAGCGCTTCCTG-3’ was used to sequence 3 variants: rs28929474 (Z), rs763023697 (Leu353Phe_fsTer24) and rs1303 (M3), primer 5’-AGCAGTGACCCAGGGATGTG-3’ was used to sequence both rs17580 (S) and rs6647 (M1(Ala213)) variants and primer 5’-AATGCATTGCCAAGGAGAGTTC-3’ was used to sequence both rs709932 (M4) and rs20546 (M3Riedenburg). Sequencing files were assembled and analyzed using the European Molecular Biology Laboratory-European Bioinformatics Institute, Clustal Omega Multiple Alignment Tool.20

Results
Sequencing and Ascertainment of Allelic Background
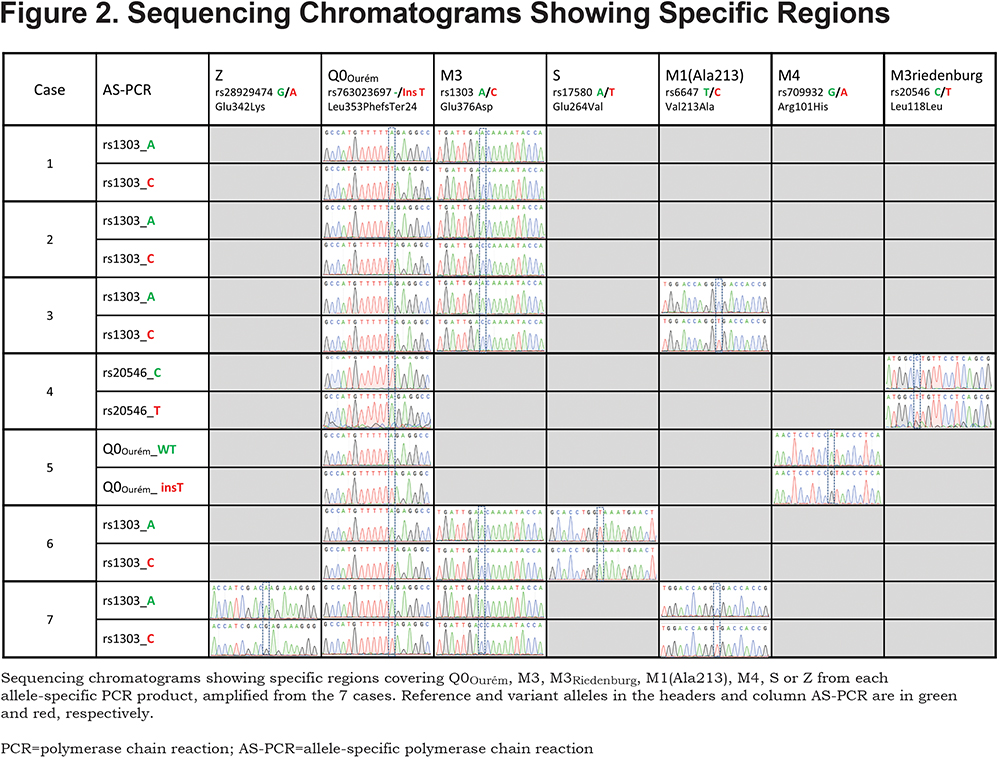
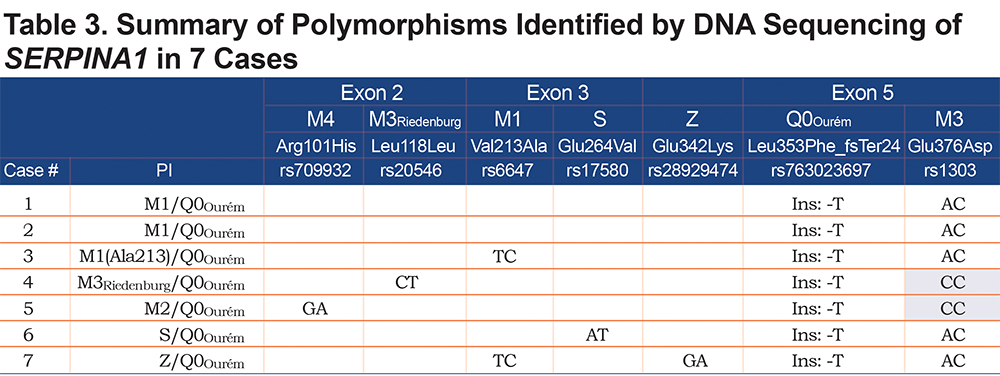
For all 7 study participants, the 4 coding exons of the SERPINA1 gene were first sequenced. Then, allele-specific sequencing was performed to determine the allelic background of Leu353Phe_fsTer24 (Figure 1).From the AS-PCR DNA sequencing data, the T-insertion (rs763023697) was found on the same allele as the M3 variant (rs1303) for all 7 individuals (Figure 2). Therefore, all 7 patients carried a Leu353Phe_fsTer24 variant on the M3 background, which corresponds to Q0Ourém on the PI system nomenclature. All 7 patients were heterozygotes for Q0Ourém with complete genotype determined as follows: 2 individuals were M1/Q0Ourém, 1 was M2/Q0Ourém, 1 was M3Riedenburg/Q0Ourém, 1 was M1(Ala213)/Q0Ourém, 1 was S/Q0Ourém and 1 was Z/Q0Ourém as schematized in Figure 3. Case 6 was carrying 1 deficient variant (S) and 1 null mutation (Q0Ourém) on 2 different alleles. Similarly, Case 7 was carrying 1 deficient variant (Z) and 1 null mutation (Q0Ourém) on 2 different alleles. More details are shown in Table 3 and Figure 2.

Clinical Characteristics of Study Participants
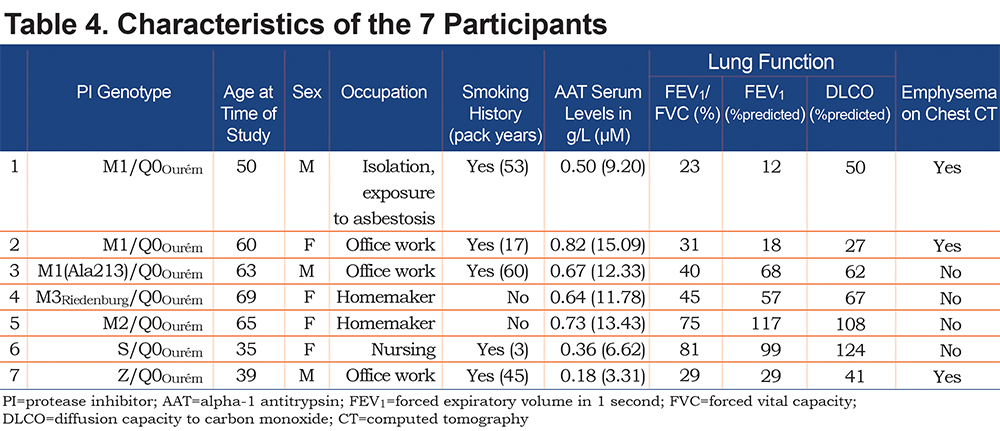
The clinical characteristics of the 7 study participants are summarized in Table 4. Three unrelated participants (cases 1, 6, and 7) were from the province of Quebec in Canada. The 4 remaining participants (cases 2, 3, 4 and 5) were from Spain. Case 1 with genotypeM1/Q0Ourém was a 50 year-old male, with a smoking history of 53 pack years, and very severe airflow obstruction associated with reduced diffusion capacity due to upper lobe predominant mixed (centrolobular and panlobular) emphysema on chest CT.Case 2 with genotypeM1/Q0Ourém was a 60 year-old female, former smoker (17 pack years), exhibiting very severe airflow obstruction and reduced diffusion capacity due to upper lobe predominant centrolobular emphysema on chest CT. Case 3 with genotypeM1(Ala213)/Q0Ourém, was a 63 year-old male, former smoker (60 pack years), with moderate airflow obstruction and reduced diffusion capacity but no emphysema on chest CT. Genotypes for cases 4 and 5wereM3Riedenburg/Q0Ourém and M2/Q0Ourém, respectively. These 69 and 65 year-old females were never smokers and had moderate airflow obstruction and reduced diffusion capacity (Case 4) or normal lung function (Case 5) and no emphysema on chest CT. Case 6 with genotype S/Q0Ourém, was a 35 year-old female, with a smoking exposure of 3 pack years, normal lung function and no abnormality on chest CT. Lastly, Case 7 with genotype Z/Q0Ourém, was a 39 year-old male, with a smoking exposure of 45 pack years who had severe airflow limitation and emphysema on chest CT. All 7 patients had reduced serum levels of AAT compared to normal values, including 3 (cases 1, 6, and 7) below the protective threshold of 0.6g/l (11mM).
Discussion
By combining our experience in 2 clinical centers, we report 7 new heterozygote patients for the null variant Leu353Phe_fsTer24 (rs763023697). In all cases, it was transmitted on the M3 background and thus corresponds to Q0Ourém. The allelic background was ascertained by using a newly developed allele-specific DNA sequencing method that consists of amplifying single alleles using primers targeting heterozygote polymorphisms and allowing us to map different variants on each copy of the SERPINA1 gene. Allele-specific sequencing was useful to clarify whether the E264V (S) and E342K (Z) observed in cases 6 and 7, respectively, were on a different allele than Leu353Phe_fsTer24.
The allelic origin of genetic variants can have important clinical meaning for patients carrying more than 1 deficient/null variants. Carrying such variants on distinct alleles increases the likelihood of developing emphysema compared to carrying 2 mutations on the same allele, a situation where the remaining allele could be fully functional. Case 6, a 35-year-old woman, had 1 deficient allele (S) and 1 null allele. Her serum AAT levels (0.36g/l) were markedly reduced and lower than the 5 Q0Ourém heterozygotes. At this young age, she did not have clinical nor CT evidence of emphysema. However, closer surveillance is warranted. Case 7, a 39 year-old man with a strong smoking history, also had 1 deficient allele (Z) and 1 null allele. He had the lowest AAT levels of all 7 patients (0.18g/l) and suffered from severe emphysema with a forced expiratory volume in 1 second (FEV1) of 29% predicted. This patient exemplifies the unfortunate and too frequent situation of AATD cases who are diagnosed late in the disease process, when severe irreversible airway obstruction and shortness of breath are already present and before being informed of their genetic predisposition to emphysema and particular susceptibility to tobacco smoke. This is an unacceptable situation as AATD can be detected at birth.
AAT levels observed in the 7 Q0Ourém heterozygotes were low, ranging from 0.18g/l to 0.82g/l (3.31 to 15.09 mM), with 3 individuals below the protective threshold of 0.6g/l. AAT levels herein are consistent with previous studies reporting this null variant and appeared to be lower than AAT levels observed in Z heterozygote patients.17,21,22 Conceivably, null heterozygote patients could have a propensity to display more severe lung disease than Z heterozygote patients. However, this needs to be balanced with the known variability in the serum AAT levels and the extent of lung disease for a given PI genotype as well as the known heterogeneity of respiratory involvement among patients with similarly low AAT protein levels.7,14,21,23-25This point can be appreciated in our study population (Table 4). For example, cases 1 and 3 have similar clinical profile, smoking history, genotype and reduced levels of AAT (0.50g/l [9.20µM] and 0.67g/l [12.33µM], respectively). However, Case 1 displays a very severe obstructive ventilatory defect (FEV1 of 12% of the predicted value) and severe emphysema on chest CT while Case 3 displays moderate obstructive ventilatory defect (FEV1 of 68% of the predicted value) and no emphysema.
The modulatory effects of tobacco smoking on the clinical expression of SERPINA1 sequence variants is also illustrated in this report where smokers and ex-smokers (cases 1, 2 and 3) exhibited more severe airflow obstruction than never smokers (cases 4 and 5). For example, Case 2, a former smoker of 17 pack years had an FEV1 of 18.2% predicted in spite of AAT levels above the protection threshold (0.82g/l). Smoking history is a well-recognized risk factor to precipitate emphysema in individuals with low AAT levels in whom tobacco consumption has been associated with a faster decline of pulmonary function and an increased mortality rate when compared to never smokers.7,23-26 However, smoking history alone does not fully explain phenotypic heterogeneity among individuals with intermediate AATD. Case 3 with a history of 60 pack years and Case 4, a never smoker, have similar lung disease severity. Note that Case 4 had a long history of asthma and had chronic eosinophilic pneumonia in 2010 that could potentially explain part of her impairment in lung function. Thus, other factors are undoubtedly coming into play to explain the phenotype including age, sex, co-existing lung conditions, occupational exposures, air pollution, respiratory infection, and uncharacterized genetic modifiers. In this small series, we have limited data to identify the specific factors contributing to the phenotype.
The prevalence of the Leu353Phe_fsTer24 mutation in the population remains uncertain, but it is likely very low. In the Canadian Cohort Obstructive Lung Disease study,27 a prospective cohort study based on random sampling of the general population across 9 urban areas in Canada, we sequenced SERPINA1 in 1359 individuals and found no carrier of Leu353Phe_fsTer24.28 This sequence variant was also absent in 2168 participants of SubPopulation in InteRmediate Outcome Measures in COPD Study (SPIROMICS), which is a multi-ethnic cohort of COPD individuals and unaffected heavy smokers (>20 pack years).29
The strength of this study was to correctly identify genotype information of patients carrying the null variant Leu353Phe_fsTer24. Not knowing the allelic background of this variant led previous case reports to incorrectly genotype Q0Ourém individuals as PI*M3Nullmattawa.17 Fortunately, this has no consequence for heterozygote individuals as PI*M3Nullmattawa (incorrect) and PI*M1Q0Ourém (correct) carry the same clinical consequences. However, for Q0Ourém carriers bearing a second deficient/null allele such as cases 6 and 7, knowing the allelic background is crucial to determining if the individual is homozygote or heterozygote for deficient/null alleles. The sequencing methods presented here can be transferred and useful to assess the allelic background of any variants (novel and rare) that cannot be inferred from other methods (i.e., isoelectric focusing, genotyping or standard DNA sequencing).
Predispositional testing is recommended by the American Thoracic Society and the European Respiratory Society and the Alpha-1 Foundation for relatives and partners of index cases.2,8,30 Knowing the specific mutations running within families is important to the study of inheritance of AATD and related liver and lung diseases. For rare cases, our study also demonstrated that allele-specific sequencing is needed. For example, the S/Q0Ourém patient (Case 6) and the Z/Q0Ourém patient (Case 7) had a 100% probability of transmitting an abnormal allele to their offspring (50% deficient, 50% null). This exemplifies how genetic counseling could be influenced by allele-specific sequencing. Allele-specific sequencing is not mentioned in AATD diagnosis guidelines. However, this method should be considered especially when discovering more than 1 SERPINA1 mutation within an individual and the allelic origin has clinical and genetic counselling implications.
To conclude, we developed an allele-specific DNA sequencing method that should be considered in rare cases to understand the clinical significance of genetic variants found in SERPINA1. The method can be particularly useful to determine the allelic background of a new mutation or to reveal if carriers of 2 pathogenic mutations have 1 or 2 defective copies of the SERPINA1 gene.
Acknowledgements
The authors would like to thank the team at the Institut universitaire de cardiologie et de pneumologie de Québec site of the Quebec Respiratory Health Network Tissue Bank for their valuable assistance.
Author Contributions: FM and YB designed and supervised the study. JB, KV, and FM recruited and performed clinical assessment of patients from Quebec City. IB, AN, and MM recruited and performed clinical assessment of patients from Barcelona. NG performed laboratory and genetic analyses. JB, NG, KV, FM, and YB performed data analysis and interpretation. JB, NG, KV, FM, and YB wrote the manuscript with input from all authors. All authors approved the final version of the manuscript.
Declaration of Interest
Marc Miravitlles has received speaker fees from AstraZeneca, Boehringer Ingelheim, Chiesi, Cipla, Menarini, Rovi, Bial, Zambon, CSL Behring, Grifols and Novartis, consulting fees from AstraZeneca, Boehringer Ingelheim, Chiesi, GlaxoSmithKline, Bial, Gebro Pharma, CSL Behring, Laboratorios Esteve, Ferrer, Mereo Biopharma, Verona Pharma, TEVA, pH Pharma, Novartis and Grifols and research grants from GlaxoSmithKline and Grifols. François Maltais received fees for speaking at conferences sponsored by Boehringer Ingelheim, Novartis and Grifols; research grants for participating in multicenter trials sponsored by GlaxoSmithKline, Boehringer Ingelheim, AstraZeneca, Sanofi, and Novartis; and unrestricted research grants from Boehringer Ingelheim, Novartis and Grifols. Yohan Bossé received speaker fees from Grifols Canada, Ltd.
All other authors have nothing to declare.