Running Head: Improved Screening for Alpha-1 Antitrypsin Deficiency
Funding Support: not applicable
Date of acceptance: November 18, 2020 ǀ Published online: December 2, 2020
Abbreviations: alpha-1 antitrypsin deficiency, AATD; chronic obstructive pulmonary disease, COPD; pulmonary function testing, PFT; odds ratio, OR; alpha-1 antitrypsin, AAT; American Thoracic Society, ATS; European Respiratory Society, ERS; Global initiative for chronic Obstructive Lung Disease, GOLD; respiratory therapists, RTs; forced expiratory volume in 1 second, FEV1
Citation: Gurevich S, Daya A, Da Silva C, Girard C, Rahaghi F. Improving screening for alpha-1 antitrypsin with direct testing in the pulmonary function testing laboratory. Chronic Obstr Pulm Dis. 2021; 8(2): 190-197. doi: http://doi.org/10.15326/jcopdf.2020.0179
Introduction
Alpha-1 antitrypsin (AAT) is a protein that has both anti-inflammatory and anti-proteolytic functions and serves to limit protease-induced injury to lung matrix proteins.1 A deficiency of AAT predisposes patients to chronic obstructive pulmonary disease (COPD) as well as other disorders including bronchiectasis and liver disease.2 Early identification of patients with alpha-1 antitrypsin deficiency (AATD) allows for interventions that can slow the effects of this disease, and improves morbidity, loss of lung function and mortality.3-6 Historically, testing of qualified patients has been extremely low.7,8
AAT functions by inhibiting neutrophil elastase, an enzyme that can damage respiratory epithelium and trigger a state of oxidative stress. By inhibiting this enzyme, AAT may serve to protect lung structure. A deficiency in AAT results in elevated levels of neutrophil elastase and increased degradation of lung tissue, with even more dramatic effects in smokers.9-12
AATD is one of the most common genetic diseases and occurs due to mutations in the SERPINA1 gene, which codes for AAT. Depending on the gene mutations, there can be a spectrum of disease severity. Those who carry at least 1 Pi Z allele, the gene that causes significant AATD pathology, are at risk. It is estimated that 2.9% of the population carries at least 1 copy of the Pi Z allele, putting them at risk for AATD. Pi ZZ is a genotype that causes severe disease and occurs in 1 in 4775 people. Pi SZ is a genotype that may also contribute to disease and occurs in 1 in 1124 patients. AATD has an estimated prevalence of about 1% to 3% of patients with COPD. It is estimated that there are at least 116 million carriers (Pi MS and Pi MZ) and 3.4 million deficiency allele combinations (PiS S, Pi SZ, and Pi ZZ) worldwide, based on population data from 2002.13 Despite such a high prevalence, AATD remains tremendously under recognized and underdiagnosed, with only 5%-10% of patients with AATD being diagnosed.6,7,14,15
According to the standards set forth by the American Thoracic Society (ATS) and the European Respiratory Society (ERS), it is recommended that all patients with incompletely reversible airflow obstruction be tested for AATD.2,16 Other respiratory authorities including the Global initiative for chronic Obstructive Lung Disease (GOLD)17 and the Alpha-1 Foundation18 have issued similar recommendations.
The rationale for these recommendations is that undiagnosed individuals lose opportunities for lifestyle modifications and therapies to help manage their disease early and reduce progression, thus, improving their quality of life and survival. Additionally, individuals with AATD who are properly diagnosed may help diagnose other family members who are similarly affected.19 Finally, targeted testing in this population has a higher diagnostic yield compared with other screening modalities.20
Presently, the standard of care is for the physician who orders pulmonary function tests (PFTs) to receive results, recognize non-reversible airway obstruction, and then instruct the patient to have a blood draw (or fingerstick or buccal swab testing if available in their office) testing for AATD genotyping. There is often a failure to screen at the provider level and noncompliance at the patient level. Prior interventions, including a “Physician Alert” in PFT reports, have yielded improvements but found that many qualified patients remained untested.21
Approaches including empowering respiratory therapists (RTs) to screen for AATD when non-reversible airway obstruction is diagnosed during PFTs improve detection rates even more. To attain this, medical directors of PFT laboratories implement a pathway whereby AATD testing is automatically offered to patients who are found to meet ATS/ERS recommendations. By increasing testing of appropriate patients, early detection of AATD and access to appropriate care can be accelerated.22,23 This requires proper education of respiratory therapy staff and proper incorporation into the workflow of the PFT laboratory.
A number of validated onsite testing kits that include the recommended genetic testing are in use and can be performed by non-physician personnel including RTs and nurses. They collect a sample from patients using a finger stick method, or a more recently available buccal mucosa swab kit method.23-25
In this study, we test the hypothesis that offering rapid, practical testing immediately on identification of at-risk patients in the PFT laboratory will result in a significantly higher percentage of at-risk patients completing screening for AATD.
Methods
Study Design and Eligibility Criteria
This is a prospective, observational study comparing the effectiveness of 4 different modalities for AATD testing. The 4 modalities include: (1) directing the patient to further discuss results with their ordering physician, (2) providing a prescription for AATD testing at their local laboratory, (3) testing via the finger stick (AlphaKit) on-site in the PFT laboratory, or (4) testing via buccal mucosa swabs (AlphaID) on-site in the PFT laboratory. Each modality was offered during a rotating bi-weekly schedule. For example, for the first 2 weeks of the study, all eligible patients were referred back to the ordering provider. During the next 2 weeks, a prescription for testing was provided, and so forth. This rotation repeated until a sample size of at least 160 patients was obtained, with at least 35 patients per testing modality. An institutional review board provided waiver of written consent.
We included patients age 18 or above with non-reversible airway obstruction detected on PFTs. These patients were identified by RTs. We excluded those patients with prior AATD testing, those with documented AATD, and those who normalized their forced expiratory volume in 1 second (FEV1) percentage with bronchodilator challenge. All eligible patients were provided with information about AATD and advised to also discuss their results with the ordering physician, including prior to AATD testing. RTs were provided with appropriate training and a script for patient education prior to the start of the study.
Outcomes and Assessments
The primary endpoint was rates of completed testing for AATD among at-risk patients as defined by the ATS/ERS. Patients who agreed to testing directly after being identified as at-risk in the PFT laboratory were provided the finger-stick blood collection or the buccal swab collection depending on the week of the study. Patients who were offered a prescription for serologic testing or who were referred back to the PFT-ordering provider were given 6 weeks to complete testing prior to being counted as non-compliant with testing. All patients were provided education about AATD. For qualified patients who did not have the testing, their reasons why were also collected.
Statistical Analyses
Univariate analysis was performed to assess the differences in baseline characteristics and screening rates between modalities in which Chi-square test, or Fisher’s exact test when appropriate, was used for categorical factors, while Wilcoxon’s rank-sum test was applied for continuous factors. Exact logistic regression model was conducted to evaluate the difference in screening rates among the groups in which the odds ratios and their 95% confidence intervals were provided. All data analyses were conducted using SAS version 9.4.
Results
Baseline Characteristics
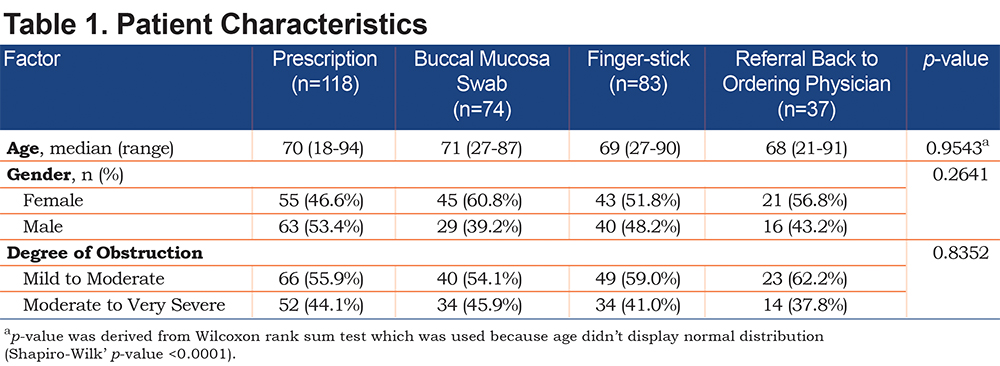
A total of 312 patients (148 [47.4%] males, and 164 [52.6%] females; median age: 70 [range 18-94 years old]) were included in the study. There was no statistical difference between the age, gender, or degree of obstruction between the patients screened with the different modalities (Table 1).
Primary Endpoints
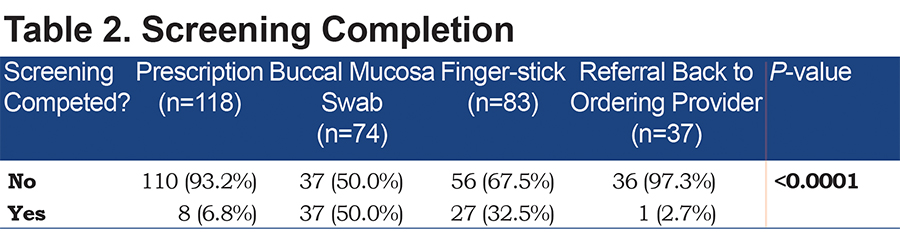
Of the 37 patients who were instructed to follow up with their ordering physician, only 1 (2.7%) completed testing. Of the 118 patients who were given a script for AAT genetic testing to be completed in a lab, only 8 (6.8%) completed testing. Of the 83 patients who were offered testing by finger-stick, 27 (32.5%) completed testing. Of the 74 patients who were offered testing via buccal mucosa swab, 37 (50%) completed testing. These differences were statistically significant with a p-value of <0.0001 (Tables 2, 3).
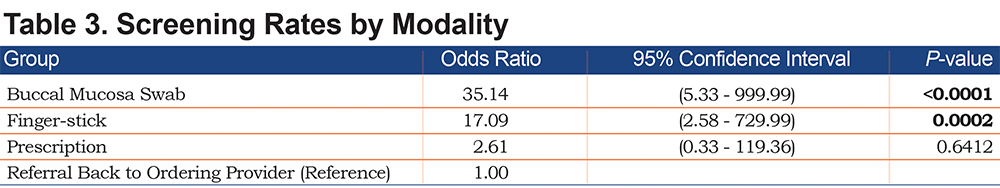
Since the current standard of practice is to have the patient follow up with their ordering physician for further discussion, this group was set as the reference group. In comparison to following up with an ordering physician, patients were 17 times more likely to be tested in the finger-stick group, and 35 times more likely to be tested in the buccal mucosa swab group.
Additional Findings
For those patients who agreed to testing, the time for RT completion averaged 5 minutes (including education) for the finger stick test, and 1 minute for the buccal swab test. A unanimous RT preference for the buccal test was reported.
When testing was indicated but not performed the most common reported reasons included the time it took to perform the test, the invasiveness of the test (for finger stick collection), and the potential concerns over collection of DNA materials in the finger stick and buccal swab tests (Table 4).

Of additional note, although this study was not powered to detect changes in testing rates, 5 carrier results (Pi MS) were identified. The fact that a deficient patient was not identified in this data set of 73 patients is not extraordinary as it estimated that you need to test 295 patients to be 95% sure to find 1 deficient patient.22
Discussion
Despite increasing knowledge of favorable outcomes in early screening of alpha-1 antitrypsin deficiency, testing continues to remain underutilized. Several studies have shown the average delay between onset of symptoms and actual diagnosis of AATD well exceeds 5 years,7,26,27 potentially resulting in deleterious effects on both a patient’s lung and liver function. According to the ATS and the ERS, screening recommendations should include those with diagnosed COPD on PFTs, bronchiectasis with no established etiology, cryptogenic liver disease, granulomatosis with polyangiitis, necrotizing panniculitis, and those with first degree relatives who are carriers of or have AATD.
There are several hypotheses as to why screening is delayed or not initiated. There is a general idea that there is inadequate awareness and education on the disease topic itself. In one study, both internal medicine residents and respiratory therapists were given a questionnaire to assess their knowledge on not only the disease but also when to screen. The percentage of correct answers was 52% and 54% in RTs and internal medicine residents respectively indicating a necessity for further education.28 Additionally, it has been questioned whether health care providers are hesitant to provide screening due to the perceived idea that there is not an effective treatment for this disease. However, a questionnaire study that focused on this idea showed that only 8% of physicians answered “there is no treatment available for this disease,” suggesting this was not a widely accepted idea.29 In addition to an increased need for education on screening and treatment of AATD, health care providers also expressed that there is a lack of feasibility in the methods for screening.
In this study, diagnosis of non-reversible airway obstruction prompted screening for AATD. Four available options were compared. Onsite testing options included having the patient submit to a finger stick blood sample collection or a buccal cheek swab sample collection. Offsite testing included a written prescription for the patient to obtain serologic AAT testing. Lastly, patients were offered the current community standard of referral back to the PFT-ordering physician to discuss further testing for AATD.
The results from this study indicate that onsite testing by far provided the highest completion rates. The buccal swab (AlphaID) had the highest completion rate of all at 50% versus 32.5% found in the finger stick (AlphaKit) method and was faster and preferred by the administering RTs. Off-site testing had the lowest completion rates - only 6.8% of patients had their blood work completed despite education and a physical prescription, and only 2.7% followed up with the physician who ordered the PFTs to obtain AATD testing.
The AlphaID buccal swab test is a minimally invasive, accurate, and rapid test that can be collected in the clinic by RTs as soon as at-risk patients are identified by the spirometry portion of their PFTs. It is currently available free to patients and providers as the cost is covered by a pharmaceutical company.30 It is important to recognize that while it remains free directly to patients getting the test, this cost is ultimately borne by the price of the therapy and the health care system.
The AlphaID buccal swab average testing time was noted as 1 minute including a brief education session. From the time the sample is taken and sent off, an average of 1 week is needed for confirmation of results. This test detects 14 genetic mutations as well as variants associated with AATD. The ease and feasibility of this method makes it ideal for both provider and patient. It is especially desirable for those who are fearful of more invasive collection (finger sticks, blood draws) or with poor adherence to follow-up visits. Those patients who did decline swab testing noted wanting to speak with their physician first, being concerned about the security of the genetic information, and equating this to commercially available broader genetic testing kits which they did not want to pursue.
The finger stick AlphaKit also had higher completion rates than those completing blood work at an outside laboratory or following up with their physician. However, this method fell short of success of the buccal swab AlphaID. The most reported reason to decline this modality of testing was the invasiveness of the procedure, as well as the extra time needed to obtain it (average of 5 minutes). Despite education to the contrary, patients were reported to equate this simple finger prick to being like that of an arterial blood gas stick or phlebotomy.
The current community standard of leaving the responsibility for AATD testing with the PFT-ordering physician yields testing rates in this study of 2%-3%, similar to those previously reported. Even with the addition of education and the provision of a written prescription, the completion rate remained woefully low. Those who did complete testing had longer wait times and more practical barriers including going to the laboratory and making sure their insurance covered the testing. Additionally, more time had to be allotted to educating the patient on the importance of getting their blood drawn and how early detection can aid in the treatment plan.
This study also re-affirms that RTs should not only be educated on this topic but also be made to feel empowered to offer AATD testing to patients who meet the criteria. It reconfirms previously demonstrated practical feasibility of testing through the PFT laboratory. This practicality very likely also carries over to desk-top spirometry testing done in many physician offices. Broader testing of these at-risk patients will ultimately help aid in early detection and treatment strategies for delaying progression of disease.
It is important to point out that there are a number of current and emerging therapies for AATD once diagnosed. From risk factor modification including smoking cessation, medical therapy for COPD, nutritional support and pulmonary rehabilitation, to supplemental oxygen when necessary, there are many interventions that become appropriate after diagnosis. Intravenous augmentation therapy to slow the progression of lung function decline, and, ultimately, lung transplant provides additional benefits.18,30
Further education and oversight of the PFT laboratory workflow, inclusion of a statement that possible AATD testing may occur at the time of PFTs in the electronic medical record for ordering physicians, COPD order sets including AATD testing, and additional time allotted for patient education may improve testing even further.
A limitation of this study is its use of a single PFT laboratory center and, therefore, may not be applicable in other testing environments. The high-volume workflow and baseline staff education also allowed for relatively simple transitions between the different testing modalities, and these resources may not be available in smaller laboratories, or as easily transitioned in larger laboratories. Another potential limitation is the COPD patient population and how representative it was of other patient communities. Of note, significantly more patients were in the “prescription” category. This was likely a result of including an additional 2-week data collection rotation as once the rotation was started, all patients in that 2-week period were counted, as well as a particularly busy 2-week block which included the opening of an additional PFT room.
Additional limitations are those inherent to time-blocked randomization. This was chosen for ease of administration and was not felt to effect randomization in any meaningful way as no other world or calendar events affected this study period. Also, to be considered is the absent equipoise between interventions. The interventions chosen are the currently available testing modalities, warranting direct comparison.
The hypothesis of this study is generally obvious but requires a paradigm shift in where, by whom, and how the sample for AATD testing is obtained. Such a change in practice benefits not only from an intellectual expectation that it will work, but data to confirm that it has worked.
Conclusions
This study reveals the significant impact of direct testing in the PFT laboratory by RTs on AATD screening rates. The availability of this testing resulted in very significant improvement in testing rates, and the less invasive and faster buccal swab test proved most acceptable to patients. The testing protocol involved minimal burden on providers.
Incorporation of this testing protocol offers a practical way to dramatically increase screening for this common and often severe genetic disorder, offering patients and their family a diagnosis and potentially bringing them to treatment at a much earlier phase of their disease.
Acknowledgements
Author contributions: FR provided the framework of the study questions. FR, SG, and AD were involved in study design. AD, CS, CG, and SG were involved in procurement of testing kits and data collection. AD was involved in the statistical analysis. All authors participated in the writing and review of the manuscript and the literature search and approved it for publication.
Declaration of Interest
Frank Rahaghi is a consultant and speaker for Grifols and Takeda. All other authors have nothing to declare.