Running Head: Variability in Serum Alpha-1 Antitrypsin Levels
Funding Support: This work was supported by local funds held by François Maltais, MD, and unrestricted grants from AstraZeneca, Boehringer-Ingelheim, Pfizer, Novartis, and Teva Pharma awarded to Ronald J. Dandurand, MD.
Date of Acceptance: August 6, 2021 | Published Online: August 13, 2021
Abbreviations: alpha-1 antitrypsin, AAT; alpha-1 antitrypsin deficiency, AATD; intraclass correlation coefficient, ICC; confidence interval, CI; World Health Organization, WHO; American Thoracic Society, ATS; European Respiratory Society, ERS; Canadian Thoracic Society, CTS; forced expiratory volume in 1 second, FEV1; forced vital capacity, FVC; body mass index, BMI; functional residual capacity, FRC; residual volume, RV; diffusing capacity of the lungs for carbon monoxide, DLCO; white blood cells, WBC; polymerase chain reaction, PCR; standard deviation, SD; AAT at the first measurement; AAT1; AAT at the second measurement, AAT2
Citation: Haillot A, Pelland A-A, Bosse Y, et al. Intraindividual variability in serum alpha-1 antitrypsin levels. Chronic Obstr Pulm Dis. 2021; 8(4): 464-473. doi: http://doi.org/10.15326/jcopdf.2021.0228
Introduction
Alpha-1 antitrypsin (AAT) is a serine proteinase inhibitor1 whose major function is to inhibit neutrophil elastase.2 When AAT is deficient, unopposed neutrophil elastase can attack fragile alveolar tissue, and the repetitive proteolytic insults eventually cause emphysema.3 Alpha-1 antitrypsin deficiency (AATD) was first described in 1963 by Laurell and Erikson.4 It is the most common cause of hereditary chronic obstructive pulmonary disease (COPD) affecting up to 5% of people with the disease when severe and intermediate AATD is included.5,6 AATD is inherited in an autosomal codominant fashion. This disorder results from mutations in the alpha-1 antitrypsin (SERPINA1) gene located2 on chromosome 14p32. Protease inhibitor M (PI*M) is the normal allele, and the 2 most frequent deficient alleles are PI*S (Glu264Val) and PI*Z (Glu342Lys).1,7
With prevalence rates ranging from 1 in 1660 individuals in Scandinavia to 1 in 3000-6000 individuals in North America,6 it was estimated in 2012 that there were at least 47.5 million carriers of different genotypes known to increase the risk of AATD-related disease, worldwide.7 Identifying this deficit is important because it should trigger smoking cessation or other environmental exposure interventions to reduce the likelihood of developing AATD-related diseases such as COPD and emphysema,8 because a specific therapy is available, and lastly because of the implications of this genetic disorder for family members of the index case.
There is some debate among international experts on who needs to be screened for AATD and how this should be done. The World Health Organization (WHO) recommends that all patients with COPD and adolescents or adults with asthma be screened for AATD using a quantitative test.9 The joint American Thoracic Society (ATS) and European Respiratory Society (ERS) statement on AATD encourages a similar approach.10 However, the Canadian Thoracic Society (CTS) proposes a more conservative strategy with targeted testing for AAT serum or plasma levels in individuals with COPD diagnosed before 65 years of age or with a smoking history of <20 pack years.6 Despite these recommendations, less than 10% of AATD individuals have been identified.11
The best approach to identifying AATD individuals is still uncertain. Diagnostic algorithms are complex and often rely on inconclusive methods. Despite recognition that the variability in AAT serum levels leads to difficulties in interpreting the results and the proposal that genotyping for the most frequent deficiency alleles should be the favored approach,12 measuring AAT serum levels is the most frequent first step when investigating AATD in symptomatic individuals.13,14 Further tests are then used if the AAT result is low.10,15 This approach may be problematic for several reasons. First, because only 70%-80% of the variation in total AAT serum concentration is attributable to the genotype,16 some heterozygous genotypes (e.g., SZ, MZ) may have AAT levels similar to those observed in MM individuals, potentially masking the underlying susceptibility to developing AATD-related disease.17-20 Second, AAT is an acute phase reactant. As such, its serum levels can rise more than 2-fold during inflammation, infection, or injury, particularly in people with intermediate deficiency.21,22 Hence, the quantitative screening test may overestimate baseline serum AAT levels and may fail to detect people with clinically significant intermediate AATD genotypes like MZ and SZ.5,23 Lastly, little information is available regarding the intraindividual variability in serum AAT level. The purpose of this study was to determine the test-retest reproducibility of AAT serum levels and to identify potential correlates of test-retest differences in AAT levels such as acute phase inflammation, time elapsed between measurements, laboratory where the measurement was done, individual characteristics, and pulmonary function data.
Methods
Study Design
We performed a retrospective analysis of a convenience sample of 255 patients with COPD who were offered AAT quantification by nephelometry following the WHO and ATS/ERS recommendations and in whom serum AAT levels were measured at least twice, on separate patient visits between 2010 and 2014, irrespective of baseline characteristics, degree of suspicion of AATD, or AAT level on the first measurement. When more than 2 measurements were available, only the first 2 were taken into consideration. All eligible patients were at least 18 years of age and had a clinically- and spirometrically-based COPD diagnosis (forced expiratory volume in 1 second [FEV1] to forced vital capacity [FVC] ratio less than 0.7). They were identified from a community respirology practice in Pointe-Claire, Québec and Cornwall, Ontario run by one investigator (RJD). To characterize the study population, relevant medical information including age, sex, body mass index (BMI), and cumulative tobacco exposure (pack years) obtained at the time of the first AAT measurement was retrieved by 2 authors (AH and AAP) between March and July 2019. Pulmonary function tests obtained at the time of the second AAT serum level determination (FEV1, FVC, FEV1/FVC, functional residual capacity [FRC], residual volume [RV], and diffusing capacity for carbon monoxide [DLCO]) were also noted. The date and laboratory where AAT serum levels were measured (Hôtel Dieu de Montréal; Centre Hospitalier Universitaire de Montréal; Hôpital Charles-Lemoyne, Longueil; Gamma Dynacare, Pointe-Claire, CML HealthCare, Mississauga and LifeLabs, Cornwall), were also noted. White blood cell (WBC) count and fibrinogen were measured at the time of the second AAT dosage in 228 and 173 patients, respectively. Among these patients, 166, 62, and 7 had both WBC count and fibrinogen, only WBC count, or only fibrinogen measured, respectively. In line with the CTS guidelines,6 genotyping for the S and Z allele was performed using polymerase chain reaction (PCR) when the AAT serum level was below 1.13g/L. Data from this cohort was retrieved from a clinical database containing anonymized demographic, clinical, and physiological data, as approved by the McGill University Health Centre Research Ethics Board (Reference number 17-198-BMA).
Statistical Analyses
AAT levels are reported ing/L, with normal values ranging from 0.9 to 2.5g/L (g/L can be converted to mg/dl by multiplying by 100). Test-retest AAT level reproducibility was assessed for the whole group and then, separately for patients who had their measurement performed in the same laboratories and those who had their measurements performed in different laboratories. Reproducibility was assessed from the intraindividual test-retest variance of serum AAT levels via calculation of intraclass correlation coefficient (ICC) considering that an ICC near 0 reflects an absence of reproducibility and an ICC of 1 indicates a perfect reproducibility.24,25 High level of concordance between test-retest AAT levels was considered when the lower limit of the 95% confidence interval (CI) of the ICC between the 2 AAT measurements26 was ≥0.75. Correlation between the 2 AAT levels was also evaluated with the Pearson correlation coefficient. We evaluated the within-individual coefficient of variation by dividing the standard deviation (SD) of the 2 AAT measurements by their mean. The limits of agreement between AAT measurements were established using Bland-Altman plots. The proportion of data points within 1.96 SDs of the mean difference between test–retest AAT difference was used to describe agreement. Pearson correlation coefficient between absolute test-retest difference and mean intraindividual values was calculated in order to detect possible influence of AAT level on the degree of agreement between the 2 measurements. As previously suggested, an AAT threshold value of 1.13g/L was used to define the requirement for further testing to establish the AATD status.22 This was based on the notion that an AAT level below this level should detect Pi*MZ, Pi*SZ, PI*MS, and PI*SS individuals with a sensitivity of 92% and specificity9,11 of 90%. The proportion of patients for whom the requirement for further testing needed to be reconsidered based on the result of the second measurements is reported in a percentage. Pearson correlation coefficients were used to identify potential determinants of the between-measurement AAT serum level variability including, time elapsed between measurements, cumulative smoking exposure (pack years), BMI, pulmonary function data, WBC count, and fibrinogen. Also, one-way analysis of variance was used to evaluate possible relationships between AAT serum level variability and sex, age at the second measurement, and whether the AAT serum measurements were done in the same laboratory or not. Data are presented as mean (±SD) and a p-value of 0.05 was used to determine statistical significance.
Results
Baseline characteristics of the study population are presented in Table 1. Mean (±SD) age was 73±8.7 years, and 47.8% of the patients were male. Patients had on average moderate airflow limitation with a mean FEV1 of 65±21 % of predicted. The mean AAT levels were 1.41±0.41g/L and 1.41±0.38g/L for the first and second measurement, respectively with a mean absolute difference between measurements of 0.34±0.39g/L. For the majority of participants (n=189; 74%), samples were analyzed in the same laboratory. Average elapsed time between measurements was 22.4+/-9.1 months (Table 2).
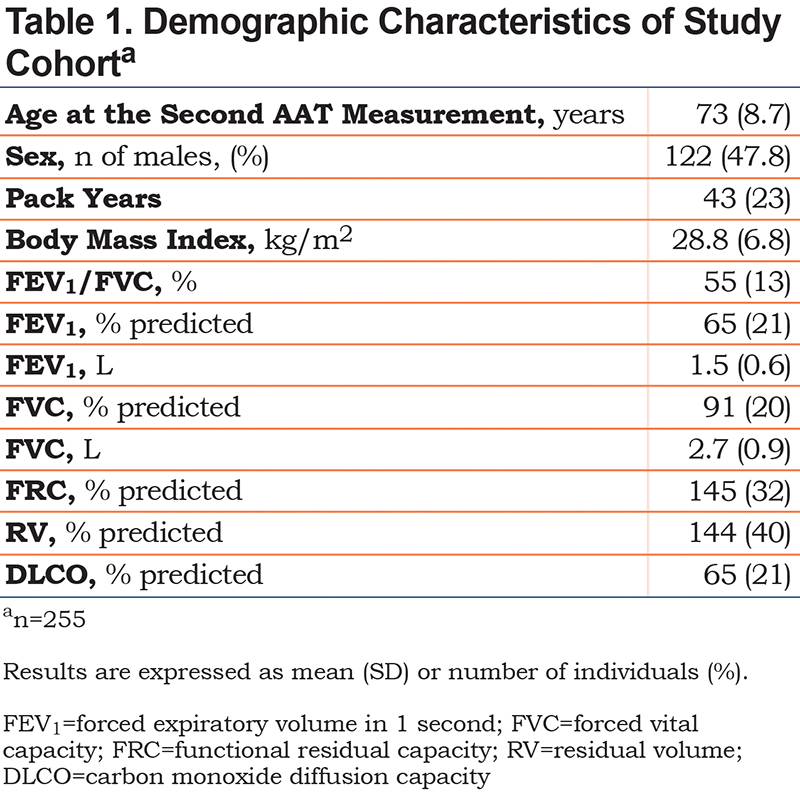
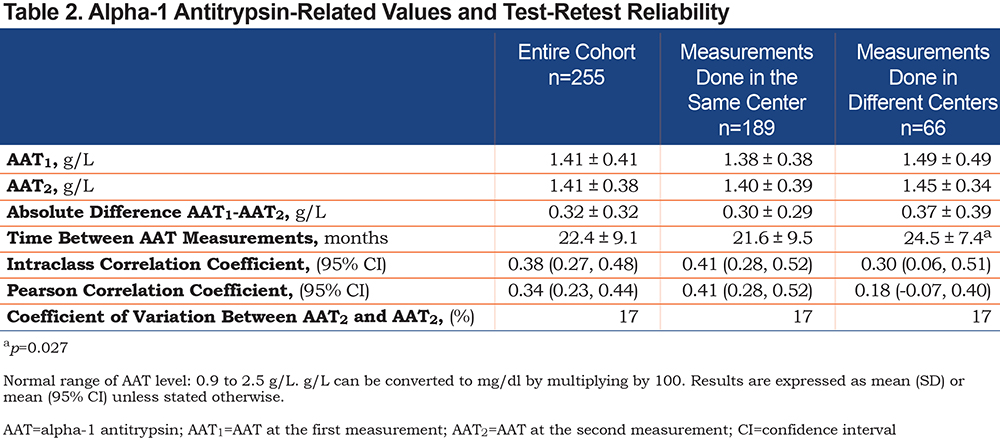
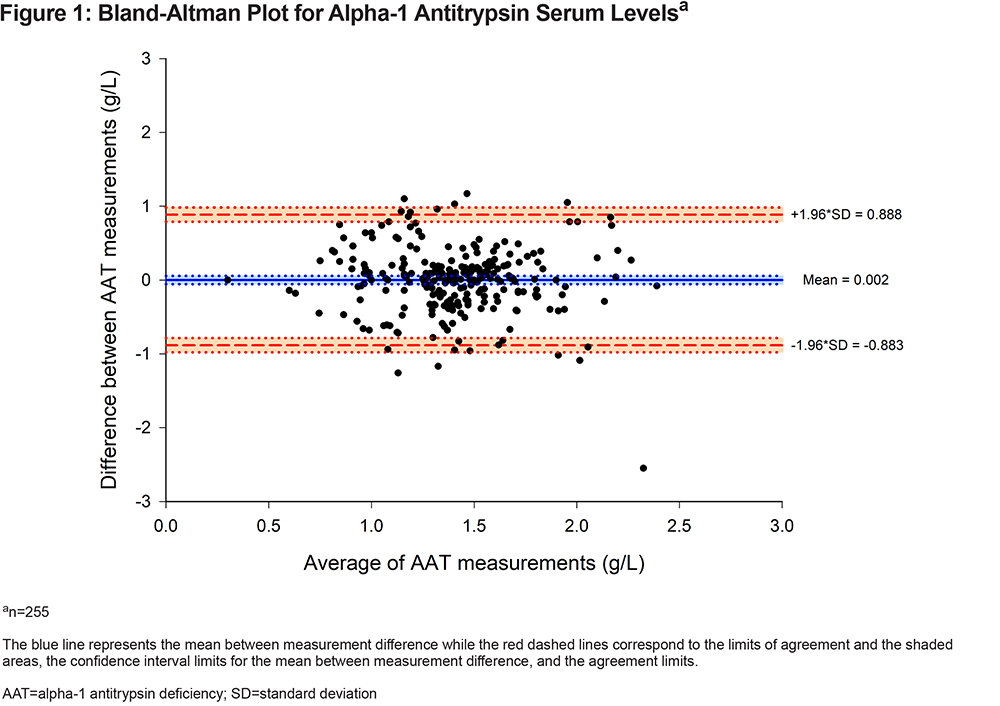
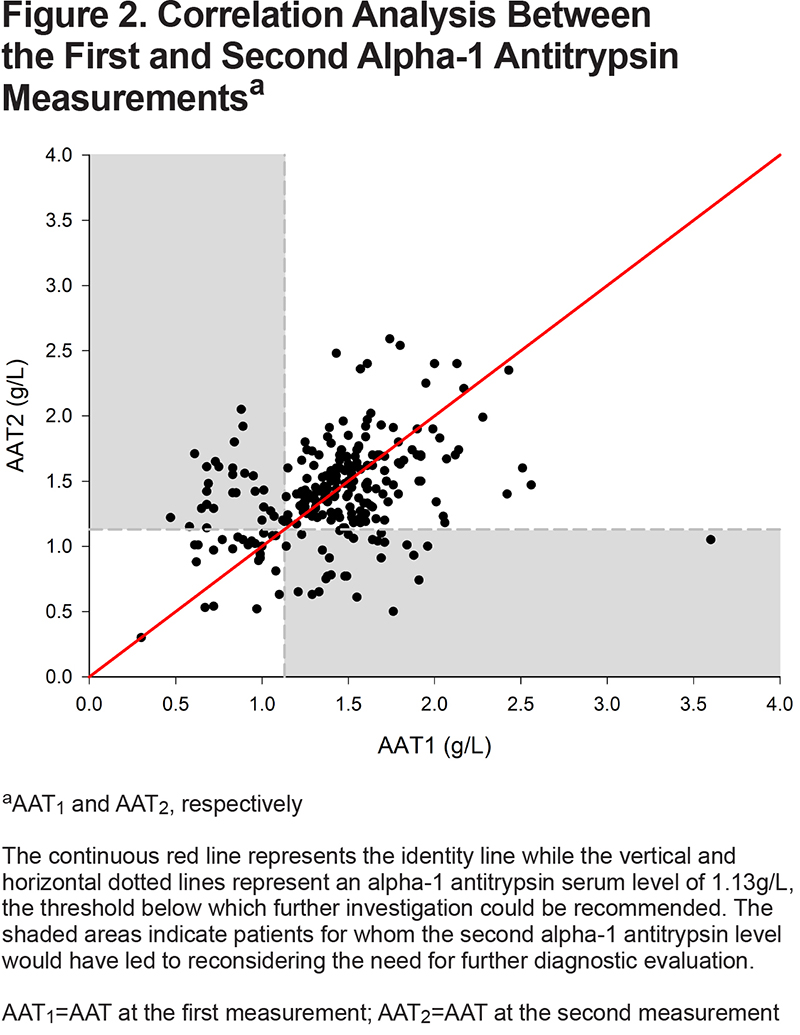
Both serum AAT levels were significantly correlated (r=0.34 [95% CI, 0.23 to 0.44], p<0.001) but the between-measurement agreement was poor as indicated by an ICC of 0.38 (95% CI, 0.27 to 0.48) (Table 2). The coefficient of variation between the 2 AAT measurements was 17%. The between-measurement agreement was similarly poor, irrespective of whether the 2 measurements were performed in the same or different laboratories (Table 2). The Bland-Altman plot (Figure 1) revealed wide 95% limits of agreement of - 0.883 to 0.888g/L between the 2 AAT measurements. No relationship was found between the mean AAT level and the between-measurement AAT variability. As illustrated in Figure 2, the second AAT level would have led to reconsidering the need for further diagnostic evaluation in 22% of patients.
WBC counts and fibrinogen levels were available in 228 and 173 patients, respectively. There was no statistical association between ∆AAT and WBC counts (r=0.05 p=0.5, n=228). However, a significant correlation was found between fibrinogen value and the second AAT level (r=0.21 p=0.004, n=173), but not with ∆AAT. The magnitude of the between-measurement differences did not relate to individual characteristics such as BMI (r=0.01, p=0.88) and cumulative smoking exposure in pack years (r =0.05, p=0.42) or pulmonary function data such as FEV1 (r=-0.08, p=0.23), FVC (r=-0.11, p=0.08), DLCO (r=-0.02, p=0.75). There was no difference in the magnitude of the between-measurement differences in AAT levels in patients who had their testing performed in the same laboratory compared to those in whom this was performed in different laboratories (Table 2). The time elapsed between measurements did not predict greater variability in AAT levels; in fact, we found the opposite, although the strength of this association was weak (r=-0.16, p=0.01).
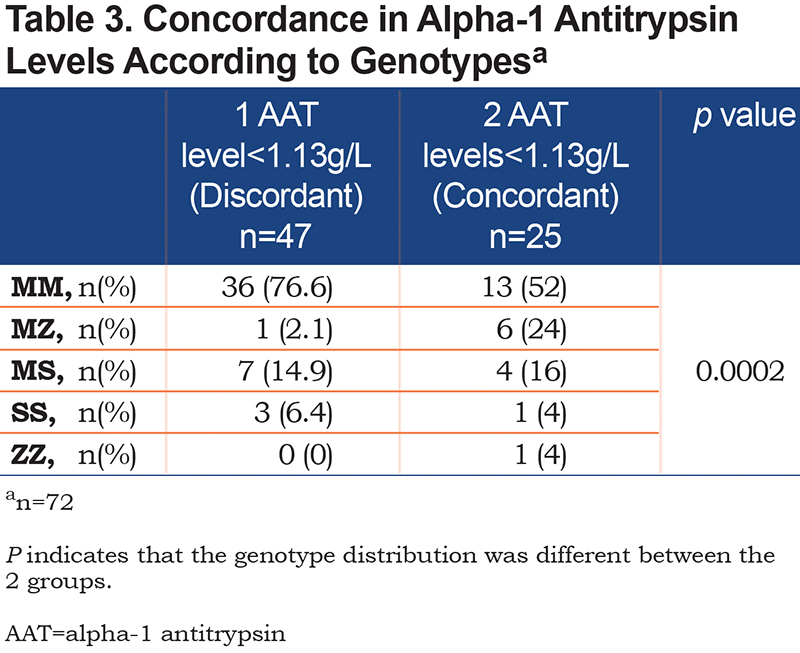
Genotyping for the S and Z alleles was available in 72 patients of the 74 patients who had at least 1 AAT serum level <1.13g/L. Overall, there were 8 clinically significant AATD genotypes detected (1 ZZ [1.4%] and 7 MZ individuals [9.7%]) in this group of patients. The distribution of genotype according to whether the AAT serum levels were discordant (i.e., only 1 value <1.13g/L, n=47) or concordant (i.e., 2 values <1.13g/L, n=25) is reported in Table 3. The genotype distribution was significantly different between the 2 groups, with a greater proportion of patients carrying a Z allele in the subgroup with concordant results <1.13g/L (p=0.002).
Discussion
This study assessed the intraindividual variations in test-retest AAT measurements, an issue that has received little attention in literature surrounding the investigation of AATD. Our results indicate that serum AAT levels showed weak intra-individual reproducibility between subsequent determinations. Furthermore, we demonstrated wide limits of agreement and a large coefficient of variation of 17% between the 2 measurements. In this cohort, discrepancies between the test-retest AAT levels would have led to reconsidering the requirement for further investigation in 22% of the individuals at the second AAT level measurement when using the 1.13g/L threshold to determine the need for additional testing. Variability in AAT serum levels could not be attributed to the laboratory where the test was performed or to the patients’ characteristics. Also, the time elapsed between measurements did not increase the variability. The observed correlation between the fibrinogen levels and the second AAT result is consistent with the notion that AAT is an acute phase reactant whose level varies in the presence of acute inflammation.5,21
Besides the well-known effects of acute inflammation on AAT levels,21,22 other factors such as tobacco smoking status,27 chronobiological fluctuations,28 and methodological differences in laboratory procedures may all contribute to the intraindividual variability in AAT levels. Irrespective of the mechanisms, this variability has implications for the investigation of AATD. We acknowledge that in patients with severe deficiency, the observed variation in AAT levels from 1 measurement to the other is unlikely to alter our conclusions; very low levels of AAT have been shown to remain relatively stable, irrespective of the testing conditions.22
The situation is less clear in patients with an intermediate deficit in whom borderline normal and even normal AAT levels are frequently found. This is not a trivial point considering that many studies have shown that MZ and SZ genotypes confer a significantly increased risk of COPD in smokers.20,29,30 Besides, intermediate deficiency is also associated with a multitude of extrapulmonary diseases such as ANCA-associated vasculitis, cirrhosis, and gallstone disease.14 We would thus contend that precise determination of AAT status is important for risk stratification of COPD and for counseling purposes even in intermediate deficiency.19,20,30 A cutoff of 1.13g/L has been previously proposed to reasonably exclude most patients with an intermediate deficiency.6,14,31 This value is based on the distribution of AAT levels in individuals with an intermediate deficit versus healthy controls and should be differentiated from the lower limit of normal for AAT serum level of 0.8 to 0.9g/L. Following previous recommendations,14,32 we genotyped patients who had at least 1 AAT level of 1.13g/L for the S and Z alleles. We found that 11% of patients who had at least 1 AAT level <1.13g/L carried 1 or 2 Z alleles. Although we do not have the corresponding results in patients ³ 1.13g/L, this 11% prevalence rate is much higher than expected in the general population.14 For example, in the CanCOLD cohort that comprises 1359 individuals who were randomly selected from the Canadian population, 3% carry a Z allele.33 We acknowledge that the 1.13g/L threshold is not universally used to direct further investigation; experts have proposed an even higher threshold10 as high as 1.4g/L, recognizing that serum AAT may reach this level, particularly during an inflammatory process.22 Using the less conservative threshold of 1.4g/L would have simply led to reassessing a larger proportion of patients to determine their AATD status with the associated cost implications of greater testing rates.
We also analyzed our data based on the concordance of the 2 AAT level measurements, a concordant result being defined as 2 AAT values above (or below) the 1.13g/L cutoff. We found that concordance in the AAT level has relevance for the underlying genotype: the proportion of patients carrying 1 or 2 deficient alleles was greater in those with 2 concordant AAT levels <1.13g/L compared to those with discordant results. Based on these observations, we would recommend further genetic testing in patients with 2 AAT values <1.13g/L. The situation is more complex for those with discordant results. Although one could argue that the distribution of AAT genotypes in this population was very similar to what is found in the general population and that no further testing is warranted, the present dataset does not allow for drawing a firm conclusion on this issue. First, all patients were tested at time of disease stability which may not always be the case when patients are tested during the course of inflammatory processes such as pneumonia or COPD exacerbation during which a 2- to 3-fold increase in AAT serum level may occur.22 The relatively small dataset also limits our ability to reach a firm conclusion on this topic. It would have been informative to have the genotype in patients above 1.13g/L for a better interpretation of the findings. Lastly, we only searched for the 2 most frequent variants (S and Z) and we may have missed rare variants with this approach. The limitations of AAT serum levels in making an accurate diagnostic of AATD were recognized in the 2016 Alpha-1 Foundation clinical practice guidelines, thus their recommendation for genetic testing as the preferred diagnostic approach in symptomatic individuals.12 Consistent with this, we recently made a similar suggestion in individuals with high suspicion of AATD (emphysema before 45 years of age, predominant basal emphysema, positive family history, and associated liver disease or bronchiectasis).34
This study highlights some limitations with the current diagnostic algorithm for AATD and it may provide some insight about why the majority of patients with AATD remains undiagnosed.11 Currently, AAT serum level testing is often the first step in the diagnostic work-up of AATD. The present results suggest that caution is warranted before confirming or excluding a diagnosis of AATD solely on the basis of AAT serum level.6,9,10 Along these lines, we submit that testing patients at the molecular level with DNA sequencing would provide complementary information regarding their AAT status and, thus, help resolve the uncertainties associated with the traditional diagnostic approach. Ultimately, a more robust investigation scheme could be instrumental in improving the rate of AATD diagnosis. Making an accurate evaluation of the AAT status has increasing clinical relevance. Patients could benefit from better medical monitoring and better prevention interventions such as a smoking cessation program8 and, in carefully selected severe AATD cases, AAT replacement therapy could be proposed. Additionally, appropriate genetic counseling relies on accurate knowledge of the AATD status. A prospective study examining the added value of SERPINA1 gene sequencing versus AAT measurement would be useful to support the hypothesis that the former approach would prove useful from a clinical standpoint and be cost effective.
Some methodological aspects of our study should be considered for a proper interpretation of the results. These considerations are linked to the retrospective nature of the present study. First, AAT analyses were sometimes carried out in different laboratories, although, the same nephelometry approach was used in all cases. Concerns could nevertheless be raised about the possibility that subtle methodological differences across laboratories could explain some of the observed variability in AAT serum levels. While this possibility cannot be fully excluded, the multivariate analysis did not reveal a contribution of the measurement location to the variability in AAT serum levels. Furthermore, the degree of agreement between the 2 measurements was similar when comparing the subgroup of patients who had their tests done in 2 different laboratories versus those in whom this was done in the same laboratories. Restricting the analysis only to those patients whose testing was done in the same laboratory (n=189) did not reduce the intraindividual variability in AAT levels. Another consideration is that the inflammatory markers (fibrinogen and WBC count) were measured only at the time of the second AAT determination and only in a subset of patients while the clinical characteristics were obtained only at the first evaluation. As such, some variations in AAT serum levels between the 2 measurements could be related to changes in clinical status (e.g., quitting smoking) and not to inflammation. It is difficult to predict how this methodological aspect of the study could have influenced our results, but, arguably, it would have been better to assess the clinical and inflammatory status simultaneously. Also, we did not systematically monitor changes in clinical status that may have occurred between the 2 AAT measurements, although both measurements were obtained during a period of apparent disease stability. Other inflammatory markers such as ferritin and C-reactive protein were not available to evaluate to the possibility of inflammation contributing to the AAT variability. Lastly, the number of cases with severely reduced AAT levels was limited, and it is uncertain to which extent the study conclusions would apply to these individuals. The present results are applicable to patients with COPD who are investigated in a community respiratory disease practice and not necessarily to other clinical situations such as liver disease or genetic counseling clinics.
Conclusion
Serum AAT levels showed weak intraindividual reproducibility between subsequent determinations. This raises concerns about the sole reliance on this parameter for AATD screening.
Acknowledgments
The authors thank Serge Simard for his help with the statistical analyses and Mickaël Martin for preparing the figures.
Author contributions:
AH, AAP, FM, and RJD were primarily responsible for the study design, analysis, interpretation, and drafting of the manuscript. YB and TPC were involved in critical review of the data and in revising the manuscript. All authors gave final approval of the version to be published.
Declaration of Interest
AH, AAP, TPC have nothing to declare. YB received speaker fees from Grifols Canada, Ltd. FM reports grants from AstraZeneca and GlaxoSmithKline, Boehringer Ingelheim, Sanofi, and Novartis, and personal fees for serving on speaker bureaus and consultation panels from GlaxoSmithKline, Grifols, and Novartis. He is financially involved with Oxynov, a company which is developing an oxygen delivery system. RJD reports grants from AstraZeneca, Boehringer-Ingelheim, Covis Pharma, Grifols, MGC Diagnostics, SCIREQ, Novartis, Pfizer, Teva Pharma, Thorasys, Vyaire, and personal fees for serving on speaker bureaus and consultation panels of Boehringer-Ingelheim, Grifols, Novartis, and Vyaire. He is President of Spiro-Tech Medical, Inc. which develops pulmonary physiology technologies.