Running Head: Nasal Microbiome and Mycobiome in COPD Patients
Funding Support: This work was supported by the National Institutes of Health’s National Institute of Environmental Health Sciences (K23ES026204, R01ES031252, P30ES000002).
Date of Acceptance: April 21, 2022 │ Published Online: April 29, 2022
Abbreviations: chronic obstructive pulmonary disease, COPD;body mass index, BMI; forced expiratory volume in 1 second, FEV1; Microbiome Multivariable Association with Linear Models, MaAsLin2; Beth Israel Deaconess Medical Center, BIDMC; ribosomal RNA, rRNA; forced vital capacity, FVC; Study of Population and COPD Exacerbations, SPACE; inhaled corticosteroids, ICSs; short-acting beta2-agonist, SABA; long-acting beta2-agonist, LABA; short-acting muscarinic antagonist, SAMA; long-acting muscarinic antagonist, LAMA; polymerase chain reaction, PCR; ribosomal internal transcribed spacer 2, ITS2; Divisive Amplicon Denoising Algorithm2, DADA2; principal coordinates analysis, PCoA; false discovery rate, FDR
Citation: Alvarez Baumgartner M, Li C, Kuntz T, et al. Differences of the nasal microbiome and mycobiome by clinical characteristics of COPD patients. Chronic Obstr Pulm Dis. 2022; 9(3): 309-324. doi: http://doi.org/10.15326/jcopdf.2021.0267
Online Supplemental Material: Read Online Supplemental Material (227KB)
Introduction
Chronic obstructive pulmonary disease (COPD) is an inflammatory lung disease that affects around 16 million Americans1 and is the third leading cause of death in the world.2 COPD is characterized by recurrent exacerbation, with half (50%) caused by bacterial pathogens, especially Streptococcus pneumoniae, Haemophilus influenzae, and Moraxella catarrhalis.3,4 These pathogens may be present in the lungs even between exacerbations.5 While studies suggest that the lung microbiome may influence the risk of COPD exacerbations, little is known about the relationship between the nasal biome and clinical characteristics or inhaled treatments of COPD patients.
The microbiomes of the upper and lower respiratory tracts are anatomically linked and share similar/overlapping characteristics.4,6,7 While the pulmonary microbiome has been highly studied, the bacterial and fungal communities of the nasal passages, which are far more accessible, have received less attention and are not well-characterized.
To address this knowledge gap, we characterized the nasal microbiome and mycobiome of both nares of 20 former smokers with COPD who were participating in a longitudinal study at Beth Israel Deaconess Medical Center (BIDMC) in Boston and agreed to participate in this nasal microbiome pilot study. We evaluated if microbial composition differed by clinical characteristics and treatments for COPD. We sampled the nasal mucosa by nasosorption, which is a less invasive, reproducible alternative to nasal lavage for the measurement of mucosal cytokines and chemokines.8 Nasosorption permits evaluation of both nasal mucosal immune responses and nasal microbial community simultaneously, using a standardized sampling approach. We hypothesized that nasal bacterial and fungal composition would differ by clinical characteristics, including COPD disease severity, symptom variability, and inhaler therapy for COPD.
Materials and Methods
Comparison of Nasosorption Versus Nasal Swab
Prior to launch of the study, we first conducted a quality control project to assess if the nasal microbial community differed when sampling the anterior nares by nasosorption (which is a more novel approach with the advantage of standardizing sample dilution)9 versus the more common approach of sampling the nares using a polyester tipped nasal swab.10,11 We compared these 2 methodologies by collecting 24 samples (sampling each nare by both nasosorption and swab) among 6 adult volunteers. All volunteers for this sampling method comparison were faculty and staff in the BIDMC Pulmonary and Critical Care Medicine division and provided verbal consent. We compared the microbial composition by 16SV4 ribosomal RNA (rRNA) sequencing of each individual, and each nare within each individual, using the 2 sampling methods bilaterally. For the nasal swab collection method, we dipped Puritan™ PurFlock™ Ultra Flocked Swabs in sterile saline and swabbed both anterior nares, as previously described.11 Then, each nare was briefly moistened with 100µl of 0.9% sterile normal saline solution. An absorbent, fibrous matrix of Leukosorb medium (Pall Scientific, Port Washington, New York), cut to fit within the nasal passages, was inserted into each nare and clamped shut with a padded nose clip for 2 min. Samples were stored in separate cryovials at -80⁰C and then shipped in bulk along with 2 control (blank) Leukosorb strips for 16SV4 rRNA sequencing at Baylor College of Medicine (Houston, Texas). We evaluated and plotted the beta diversity of the 24 samples by principal coordinate analysis ordination. We conducted a PERMANOVA analysis to assess whether left versus right nare (to assess the within-person consistency), methods of nasal microbiome collection, and participant predicted microbial community composition. This analysis was performed using the adonis function from the vegan package in R.12
Study Population
The study population consists of 20 former smokers with a COPD diagnosis, a ≥10 pack-year smoking history, and at least moderate Global initiative for chronic Obstructive Lung Disease (GOLD)13 stage 2 airflow (defined as forced expiratory volume in 1 second [FEV1]/forced vital capacity [FVC] <70% and FEV1 <80% normal) obstruction on spirometry. All participants were originally recruited as part of the Study of Pollution and COPD Exacerbation (SPACE) at BIDMC in Boston, Massachusetts.14 Exclusion criteria included smokers in the household, history of lung cancer, interstitial lung disease, or bronchiectasis. All participants provided written consent to participate in both the SPACE study and the nasal microbiome pilot study, and the study protocols were approved by the institutional review board at BIDMC.
Clinical Information
Demographic and clinical characteristics were collected from each participant at the BIDMC clinical research center as part of the SPACE study between February 24, 2017, and January 17, 2019. Lung function was measured at the BIDMC clinical pulmonary function laboratory according to American Thoracic Society standards, using National Health and Nutrition Examination Survey prediction formulas.15 A clinical research nurse measured vital signs, including body mass index (BMI), and determined baseline supplemental oxygen and inhaler use, including inhaled corticosteroids (ICSs), and short- and long-acting beta2-agonists (SABA and LABA) and anti-cholinergic inhalers—short-acting muscarinic antagonist (SAMA) and long-acting muscarinic antagonists (LAMA). Participants self-reported age, sex, race, and smoking history by questionnaire. As part of the SPACE study, participants were then followed prospectively for a total of 4 non-consecutive observation months (1 month per season) for 1 year. During follow-up, they completed a validated daily symptom questionnaire16,17 to document changes in respiratory symptoms. Participants were categorized as having high versus low symptom frequency based on reporting ≥10 versus <10 days of worse-than-baseline respiratory symptoms (including dyspnea, sputum quantity or color, cough, wheeze, chest tightness, nasal congestion, sore throat, or fever) during the 4 months of follow-up. The study protocol is illustrated in Figure 1.
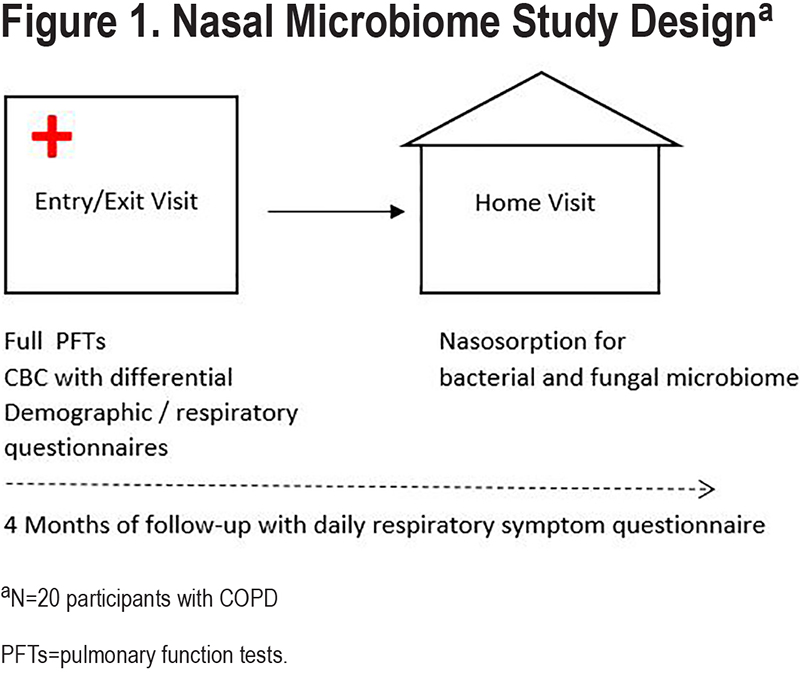
Nasal Sample Collection and Sequencing
Nasosorption was collected for bacterial and fungal microbiome at a home visit within weeks of the clinical visit at BIDMC (Figure 1). Home visits occurred during a period of stable COPD for all participants, and visits were rescheduled if the participant reported symptoms concerning for an acute exacerbation. As described above, each nare was briefly moistened with 100µl of 0.9% sterile normal saline solution and an absorbent, fibrous matrix of Leukosorb medium (Pall Scientific, Port Washington, New York), cut to fit within the nasal passages,8 was inserted into each nare and clamped shut with a padded nose clip for 2 min. All 40 paired samples plus 4 control Leukosorb strips were stored at -80ᴼC and subsequently underwent DNA extraction. Total genomic DNA was extracted using the Qiagen MagAttract PowerSoil DNA Kit (formerly sold by MO BIO as PowerMag Soil DNA Isolation Kit). The 16S rRNA region was amplified by polymerase chain reaction (PCR) and sequenced on the Illumina MiSeq platform. The primers used for amplification contained adapters for MiSeq sequencing and single-index barcodes so that the polymerase chain reaction (PCR) products could be pooled and sequenced directly, targeting at least 10,000 reads per sample.18 16S rRNA sequencing was conducted with 515F and 806R primers (2x250 bp paired-end), ribosomal internal transcribed spacer 2 (ITS2) sequencing was conducted with ITS3F and ITS4R primers (2x300 bp paired-end).19
Bioinformatics
Raw fastq amplicon sequences were bioinformatically processed by the Harvard T.H. Chan School of Public Health Microbiome Analysis Core through the Divisive Amplicon Denoising Algorithm 2 (DADA2)20 workflow in R which has been wrapped in the reproducible bioBakery workflow21 with AnADAMA2. Briefly, sequences are demultiplexed and then DADA2 uses R scripts to denoise, filter, trim, merge, remove chimeras, and taxonomically classify paired-end Illumina sequence data. DADA2 assesses the reads for sequence error rates and corrects on a base-by-base basis. Chimera removal occurs while simultaneously assigning amplicons to amplicon sequence variants. Phylogenetic trees are constructed after alignment of sequences using Clustal Omega.22 Amplicon sequence variants are then taxonomically assigned at the genus level using the SILVA23 reference database with 97% identity threshold for bacterial taxa and UNITE reference database for fungal taxa.24 For functional profiling, the PICRUSt225 algorithm infers a metagenome, the gene content, and gene abundance, from the community taxonomies; based on phylogenetic proximity of a 16S-identified taxon to its closest fully sequenced relative.
Quantitative Analyses
Downstream visualization and statistics on the taxonomic abundance data were also performed. The feature table was filtered requiring all taxa to be at least 10% prevalent and have at least 0.01% abundance. Beta diversity, a measure of between-sample diversity, was calculated using the vegdist function from the vegan package in R. The Bray-Curtis dissimilarity is an index of beta diversity which considers the relative prevalence and abundance of each clade between samples. The maximum percentage compositional change (R2) associated with each clinical covariate was calculated using an omnibus univariable PERMANOVA test on the Bray-Curtis dissimilarities. Additionally, multivariable models were conducted using a PERMANOVA test on the Bray-Curtis dissimilarities with the adonis function within the vegan package in R, with permutations performed blocked within participant to account for repeated measurements (left and right nares). Further, principal coordinates analysis (PCoA) plots were created on the Bray-Curtis dissimilarities. To display the samples’ community compositions, stacked bar plots and heat maps on taxonomic relative abundances were constructed on the top 29 and 30 taxa, respectively, and annotated with the metadata. All diversity trends and community composition visualizations were created using the ggplot2 package in R.12,26
After establishing the global trend in the microbiome over metadata, per feature associations between the nasal bacterial and fungal compositions and metadata were identified using Microbiome Multivariable Association with Linear Models (MaAsLin2) package (v 1.4.0).27 MaAsLin2 employs mixed linear models to calculate all features’ associations with metadata. Filtered relative abundance normalized data is log-transformed before testing. Among each of the comparisons generated, multiple comparisons are adjusted using the Benjamini and Hochberg correction and false discovery rate (FDR) corrected p-values of 0.1 or lower are reported as significant. Thus, MaAsLin2 identifies microbial organisms that reach a statistically significant association with each of the metadata variables.
The following metadata were evaluated: age, sex, BMI (≥ versus < 30 kg/m2), FEV1 (≥ versus <50% predicted), supplemental oxygen use, inhaler use (ICS, LABA, SABA, LAMA and SAMA), nasal steroid use, high versus low COPD symptom exacerbation frequency (as defined above), lower versus higher baseline peripheral eosinophil level (< versus >150 cells/µL),28 and calendar season of nasal sample collection. We selected these clinical characteristics based on biologic plausibility (e.g., inhaled medication use) and/or prior publications suggesting an association with differences in the microbial composition of the respiratory tract.29-36
Comparison of nasal samples with the 4 blank samples confirmed (eFigure1 in the online supplement) that among top 30 genera, the majority of taxa were detected in nasal samples only. Few taxa detected in both blanks and nasal samples (for example, Corynebacterium, Staphylococcus)37 were determined to be commensal bacterial in nasal cavity and/or biologically plausible to be present in nasal samples, therefore, were kept in downstream analysis. The results of the literature review of the genera present on blanks and nasal samples are reported in the eTable 1 in the online supplement.
Results
Nasosorption Versus Nasal Swab Collection Method
In the quality control assessment of 6 adult volunteers, bacterial microbial community composition differed significantly by individual (p=0.001), but not by nare or collection method. In the PERMANOVA on the Bray-Curtis dissimilarity, individual participant explained 67.6% of the variability in microbial community composition (p=0.001), but nare (left versus right, R2=0.05, p=0.894) and sampling method (R2=0.015, p=0.366) did not. The PCoA analysis on the Bray-Curtis dissimilarities (eFigure 2 in the online supplement) showed that the bacterial composition of nasal samples collected from the same individual were very similar to each other, regardless of collection method (swab versus nasosorption).
Study Participant Characteristics
Details of the study participants are listed in Table 1. Half of the participants were 70 years of age and older (50%) with a mean age of 71±7.8. A slight majority (60%) were female, and the mean pack-year tobacco history was 51 years. Lung function was severely compromised. Half of the patients had an FEV1 <50% predicted and 35% of participants used supplemental oxygen. A majority of participants used ICS (55%), SABA (80%), LABA (60%), and LAMA (55%) inhaler treatment (alone or in combination), while only 15% used a SAMA inhaler and 15% used nasal steroids routinely.

Nasal Bacterial Microbiome
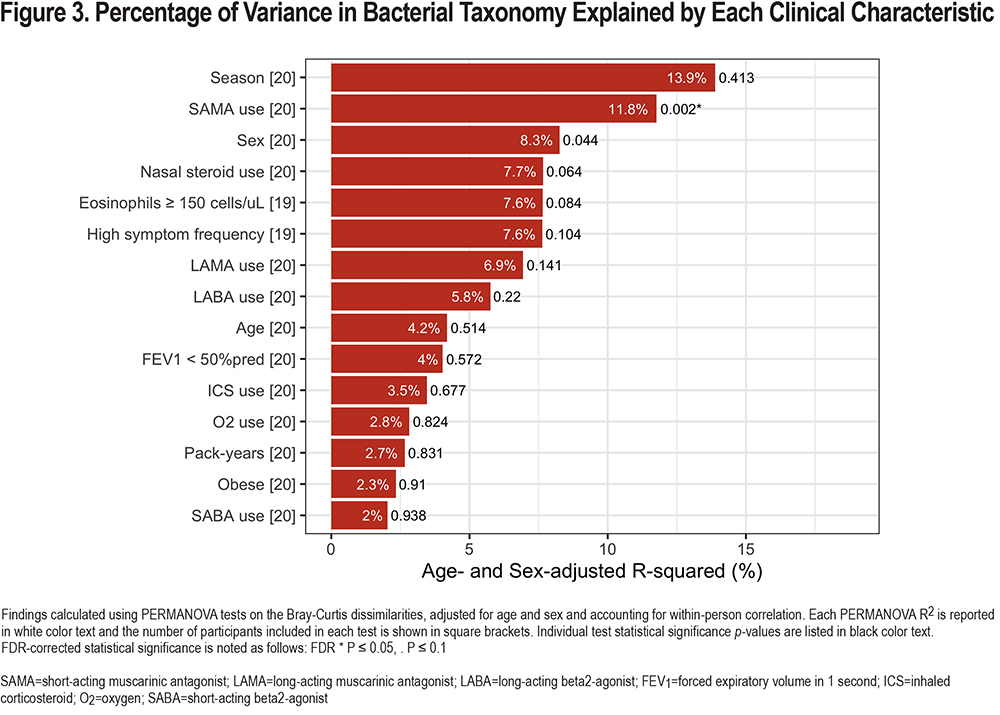
After abundance and prevalence filtering (details in the Materials and Methods section), a total of 168 different genera were detected in the bacterial community composition of participants’ nasal samples. First, to examine the overall distribution of the different genera, we plotted the top 29 taxa among all 40 participant samples collected (including the results of both nares from each participant, Figure 2), and found that the 5 most common genera were Corynebacterium, Staphylococcus, Streptococcus, Moraxella, and Dolosigranulum. We then performed a PERMANOVA test on the Bray-Curtis dissimilarities to calculatethe proportion of variability in the nasal bacterial (16S rRNA) community composition that was explained by each clinical characteristic in multivariable models adjusted for age and sex (Figure 3). While not a statistically significant predictor of community composition, the calendar season of sampling explained the highest proportion of variability (13.9%) of all characteristics examined. SAMA use explained 11.8% of the variance (p=0.002), sex (p=0.044) 8.3% of the variance, nasal steroid use (p=0.064) 7.7% of the variance, and higher eosinophil level (p=0.084) 7.6% of the variance. Of these, only the association with SAMA use met FDR-corrected statistical significance. Results of the unadjusted models were similar and are shown in eFigure 3 in the online supplement.
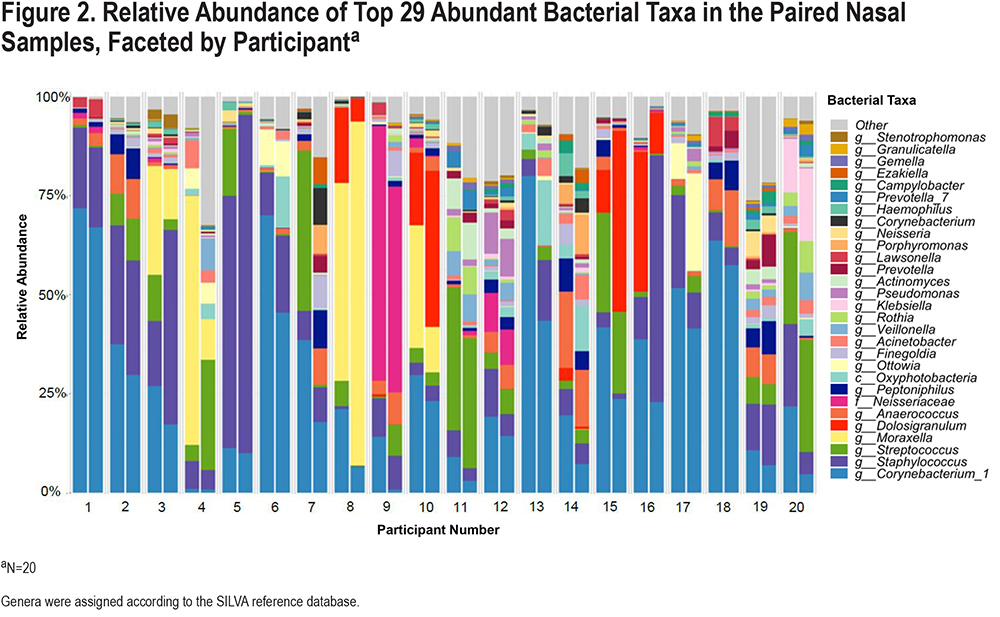
Next, we plotted the top 29 bacterial taxa according to the clinical characteristics of SAMA use, eosinophil level, nasal steroid use, and sex (Figure 4). We found that among SAMA users, there was a higher relative abundance of Moraxella (35% versus 2.4%) and Neisseriaceae (19.4% versus 0.8%), while among non-SAMA users, the top 2 genera were Staphylococcus (17.7% versus 5.1%) and Corynebacterium (31.7% versus 7.3%). Women had a higher relative abundance of Staphylococcus (21.9%) compared to men (11.8%). Nasal steroid-users had a higher relative abundance of Staphylococcus (29.9% versus 13.4%) and Neisseriaceae (19.4% versus 0.8 %) and a lower relative abundance of Corynebacterium (6.3% versus 31.9%) in the nasal fluid. Among participants with a peripheral eosinophil count ≥150cells/µL, the relative abundance of Corynebacterium was higher (42%) than in those with a lower eosinophil level (20%). Staphylococcus was more abundant in the high eosinophil group (22.8% versus 12.7%), while Moraxella was more abundant in the lower eosinophil group (10.4% versus <1%). None of these clinical characteristics were significantly associated with the relative abundance of bacterial taxa in feature-wise analyses using MaAsLin2.

Nasal Fungal Mycobiome
We compared fungal taxa that were detected in the 4 blank samples and the participant nasal samples, and found that among the top 30 genera, the majority of taxa were detected in the nasal samples only (eFigure 4 in the online supplement). After abundance and prevalence filtering (details in Materials and Methods section), a total of 69 different genera were detected in the fungal community composition of participants’ nasal samples. Similar to our analyses for the nasal bacterial microbiome, we plotted the relative abundances of the top 29 taxa among all 40 participant samples collected (Figure 5) and found that the 5 most common genera were Malassezia, Candida, Malasseziales, Debaryomyces, and Didymosphaeriaceae. We then calculatedthe proportion of variability in the nasal fungal (ITS2) community composition explained by each clinical characteristic in multivariable models adjusted for age and sex (Figure 6). Eosinophil level explained 14.4% of the fungal community variance (p=0.007). While not statistically significant (P=0.098), low FEV1 explained 7.5% of the variance and age explained 7.2% of the variance. Of these, only eosinophil level met FDR-corrected statistical significance. Results of the unadjusted models were similar and are shown in eFigure 5 in the online supplement.
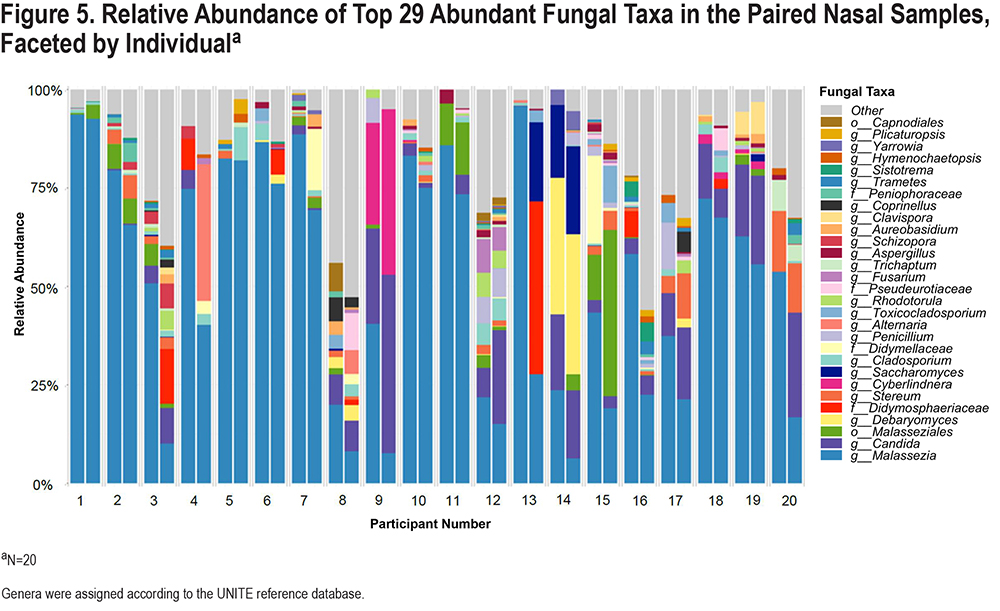
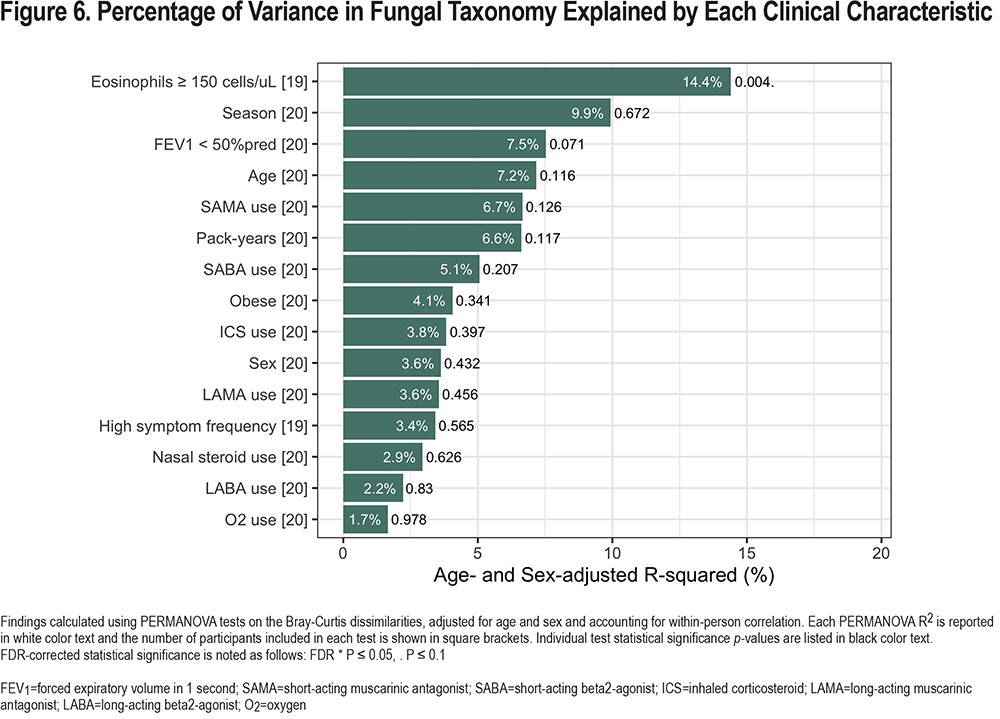
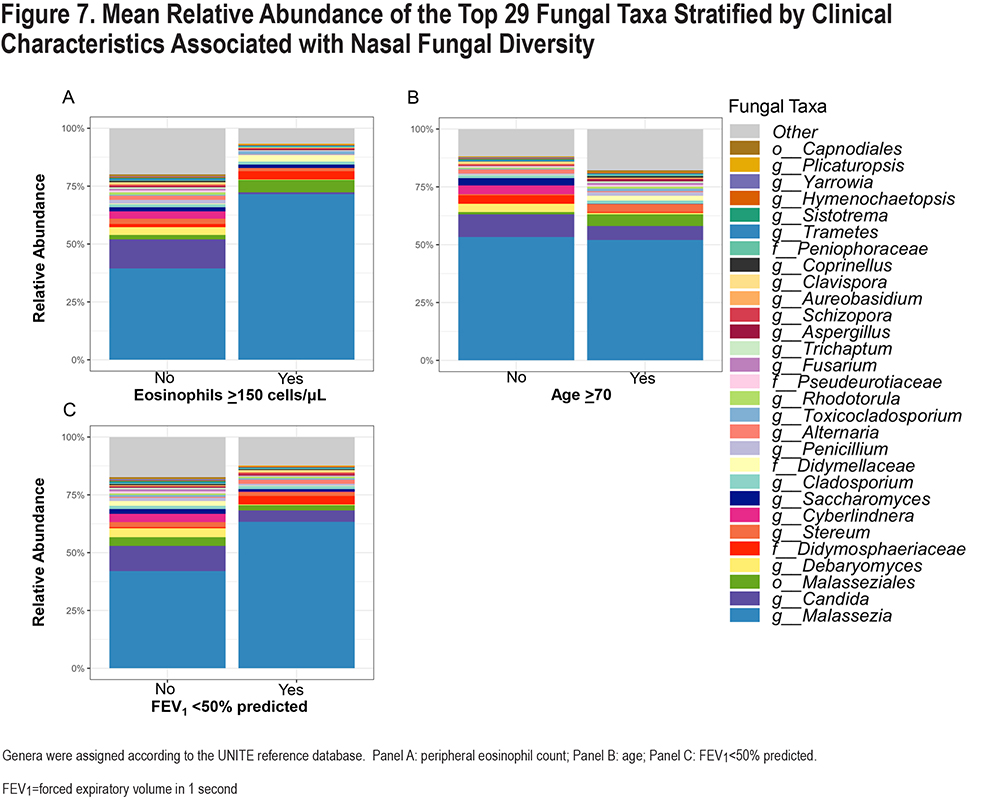
We then plotted the top 29 fungal taxa according to eosinophil level, low FEV1, and dichotomized age group (age ≥ versus < 70) (Figure 7). We found that among those with a higher eosinophil level, the relative abundance of Malassezia was higher (71.6%) than those without (39.4%), and the abundance of Candida was lower (0.7% versus 12.6%) than in non-eosinophilic patients. Among those with severely reduced FEV1 (<50% predicted), a similar pattern was observed: higher Malassezia (63.3% versus 42%) and lower Candida (4.9% versus 10.9%) than those with less impaired lung function. None of these clinical characteristics were significantly associated with the abundance of fungal taxa in MaAsLin2 analyses.
Discussion
In this small study of 20 patients with moderate-to-severe COPD, we found that the nasal bacterial and fungal communities differ between individuals, and that microbial composition may be explained, in part, by clinical characteristics of COPD patients. For the bacterial nasal microbiome, the most abundant taxa differed between individuals with COPD, whose nasal fluid was variably dominated by Corynebacterium, Moraxella, Staphylococcus, Neisseria, and Streptococcus genera. SAMA use, though relatively uncommon in this study, was significantly associated with bacterial community composition, even after FDR-correction. The composition of the nasal bacterial microbiome also differed by sex, high COPD symptom frequency, and blood eosinophil level. The nasal mycobiome was more consistently dominated by Malassezia and Candida genera, although, some individuals had substantial proportions of other genera, such as Cyberlindnera and Debaryomyces. The nasal mycobiome differed by blood eosinophil level, low FEV1, and age. While no causal inferences can be made from this cross-sectional evaluation of only 20 patients, our findings might inform future prospective research on the determinants of microbial communities in the upper respiratory tract.
Inhaler medications deposit directly into the airways, and therefore, it is conceivable that these medications would influence the composition of microbial communities lining the respiratory epithelium. There has been particular interest in evaluating the influence of inhaled ICSs on the respiratory microbiome, because ICSs suppress mucosal immune responses to microbes, and thereby, increase risk of bacterial pneumonia and oral candidiasis.38-40 In the stable state, ICS use by COPD patients has been associated with a higher bacterial load of potentially pathogenic bacteria (Haemophilus influenzae, Streptococcus pneumoniae, Moraxella catarrhalis), as measured by quantitative PCR,41 which may explain the link between ICS use and risk of bacterial pneumonia. Few studies have evaluated how ICSs, nasal steroids, and other inhaled medications might affect the composition of bacterial and fungal communities lining the respiratory tract in the stable state. While most prior work has focused on the relationship between inhaled medications (especially ICSs) and the microbiome of the lower respiratory tract, studies have shown that the microbial compositions of the upper and lower respiratory tract are highly correlated. The nasal cavity and nasopharynx are known to act as reservoirs for potential respiratory pathogens before spreading into the lower airways,42 suggesting a potential anatomic mechanism for the effect of inhaled medication use on the nasal microbiome. This is further supported by evidence showing that inhalation of cigarette smoke also influences the nasal microbiome.43 A recently published study involving 15 COPD patients who provided sputum, oral, and nasal microbiome samples found clustering of beta diversity dissimilarity by ICS use, suggesting that ICS use may shift the composition of the respiratory (including nasal) microbiome.44 A study analyzing the bronchoalveolar lavage fluid of 22 moderate-to-severe COPD patients found that the bacterial composition of the lavage fluid of those using ICSs, and also those using inhaled bronchodilators, were more alike.36 However, a much larger study that analyzed the bronchoalveolar lavage fluid of 181 people enrolled in the SPIROMICS study, including 78 with COPD, found no association between ICS use and the respiratory microbiome.45 The authors attributed the lack of an association to the relatively low prevalence of ICS use at the time of bronchoalveolar lavage. We also did not find an association between ICS use, which was common in our study (55% of participants), and bacterial or fungal composition. However, the use of intra-nasal steroid medication (by only 15% of participants) was associated with nasal bacterial community composition (p=0.064), and there was a higher proportion of potentially pathogenic Staphylococcus and Neisseriaceae among nasal steroid users and lower proportion of benign Corynebacterium (though none of these associations reached statistical significance).
SAMA use, which was also relatively rare (15%), explained 12% of the variance in bacterial taxonomy. A growing body of evidence suggests that interactions between drugs and bacteria are common.46 While we are not aware of any prior studies directly examining the effect of SAMA on the nasal microbiome, in an animal model the use of ipratropium reduced inflammatory mediators and neutrophilic infiltration in the respiratory tract,47 and therefore, our finding that SAMA use is associated with a difference in the nasal microbiome is biologically plausible. Among SAMA users, the potentially pathogenic Moraxella was the most abundant bacterial taxon, while it contributed relatively little to the nasal bacterial composition of non-SAMA users. Even though the association between SAMA use and bacterial composition reached FDR-corrected statistical significance, this may be a chance finding, particularly in light of the very small number of participants using SAMA in our sample, and will require replication, ideally in the context of a prospective study assigning inhaler treatment. Most of our participants used multiple inhalers, and there was significant overlap in use of these medications. We tested each inhaler category as a separate predictor of the nasal biome and could not evaluate the independent effects of each inhaler type, due to the limitations of our study size and design.
Studies have suggested that the microbiome of the lower respiratory tract largely is determined by the microbial community of the upper respiratory tract,36,48 and that persistence of potentially pathogenic bacteria in the airways increases risk of COPD symptoms and exacerbations.49 We evaluated if the composition of the upper airway (nasal) microbiome differed based on the frequency of worsening COPD symptoms, as determined by 4 months of daily respiratory symptom survey completion. We found that the nasal microbiome differed based on symptom frequency, although, we did not find clear evidence that the proportion of potentially pathogenic taxa was higher in the high symptom frequency group. For example, those with higher symptom frequency had a higher proportion of Corynebacterium (which is not pathogenic) and Staphylococcus, and a lower proportion of Streptococcus and Moraxella genera compared to the lower symptom frequency group. We did not find that the composition of the nasal fungal communities differed by frequency of symptom exacerbation.
Some studies involving patients with chronic lung disease, especially asthma, have found that the severity of lung function impairment is associated with differences in the bacterial microbiome.4,50,51 We compared the nasal bacterial and fungal communities among those with severe (i.e., FEV1<50% predicted) versus moderately severe airflow obstruction and found no evidence to suggest that the bacterial microbiome varied by FEV1. Other studies of the respiratory microbiome of COPD patients have also not detected differences by FEV1,44,45 which may be due to the fact that these individuals with COPD all already have moderate-to-severe lung function impairment, and further differences in microbial composition may be explained by other factors. We found that nasal fungal composition differed among those with severely impaired FEV1, with a higher proportion of Malassezia genus and a lower proportion of Candida in the lower FEV1 group.
We found associations between both the bacterial and fungal nasal microbial composition and blood eosinophil level (≥ versus. <150cells/µL). Patients with a higher eosinophil level had a lower abundance of Moraxella, a common pathogen in COPD exacerbations, but a higher abundance of Staphylococcus, which can also be pathogenic.35,52 A growing literature suggests that eosinophilic COPD patients (commonly defined as a blood eosinophil count >150cells/µL or >2%) may be less likely to experience exacerbations due to bacterial pathogens.53 Some studies have suggested that the airway bacterial load of eosinophilic COPD patients may be more stable, and less likely to be modified by suppression of the airway immune system. For example, in a randomized controlled trial of patients with moderate COPD, long-term treatment with ICSs resulted in a higher bacterial load in sputum, but only among those without blood eosinophilia.54 We found that Moraxella was less common in patients with higher blood eosinophil levels, and also less common among those higher symptom exacerbations during 4 months of observation. This suggests that baseline Moraxella may not be a determinant of COPD symptom exacerbations, at least in this small patient population of whom half had a peripheral eosinophil count ≥150cells/µL.
When examining the fungal biome, we found that patients with a higher eosinophil level had a higher abundance of Malassezia genus, while patients with a lower level had a higher relative abundance of Candida. The significance of these findings is unclear. Little is known about the effect of upper airway fungal communities on COPD exacerbations, although a recent multi-center longitudinal study of COPD patients found that airway mycobiome dominated by Saccharomyces was associated with increased COPD symptoms (by COPD Assessment Test score), and airway Aspergillus, Curvularia, and Penicillium (and specific IgE to these fungi) was associated with very frequent COPD exacerbations and higher mortality.55 It is well-established that COPD patients with eosinophilia are more likely to experience non-infectious (i.e., non-bacterial, non-viral) COPD exacerbations that are characterized by eosinophilic airway inflammation.53 Future research is needed to determine if fungal communities influence symptoms or airway inflammation in eosinophilic COPD.
We found sex-specific differences in the nasal microbiome of COPD patients, characterized by a higher abundance of Staphylococcus among women. It has been proposed that differences in the composition of the airway microbiome of men and women, determined by host factors such as sex hormones, anatomy, and the immune environment, may explain some of the sex differences in the clinical features of COPD.56 For example, women tend to develop COPD at an earlier age than men, and tend to have better lung function but more dyspnea and more frequent COPD exacerbations than men.57,58 So far, little has been reported on sex differences in the lung microbiome in COPD. In mice, the lung microbiome has been found to vary by sex, even after controlling for diet and environmental exposures.59 Among mice whose diet was supplemented with Vitamin D, the lungs of female mice were found to have a higher proportion of Acinetobacter, which may help protect against allergy.60 In females with cystic fibrosis, the proportion of mucoid Pseudomonas aeruginosa bacteria has been found to vary with estradiol levels, suggesting an influence of sex hormones on the airway microbiome,61 while males with cystic fibrosis have been found to have a more diverse respiratory microbiome than females, with higher abundance of Streptococcus and Stenotrophomonas and a lower abundance of Pseudomonas.62The Human Microbiome Project found that women had greater species diversity in the anterior nares than men, and noted differences in the species composition of the nasal, skin, and gut microbiome by sex.63 There is a need for future research that evaluates whether these apparent sex-based differences in the respiratory microbiome explain differences in the onset and clinical manifestations of COPD among women compared to men, and whether sex-based hormonal or immune interactions with the respiratory microbiome can be modified to improve the clinical course of COPD.56
In this study population which included only older adults, we did not find that the bacterial microbiome differed by age. Although we found some evidence that the nasal fungal biome differed by age, the genus-specific differences were not striking (e.g., Candida and Malassezia were the dominant genus in the nasal fluid of those > versus < 70 years of age). Overall, given the fairly narrow range of ages included in our sample, we are unable to draw firm conclusions about whether age is associated with differences in the nasal microbiome. We also did not find that the biome differed by supplemental oxygen use. This was a somewhat surprising result, since oxygen is delivered directly into the nasal passages. It is biologically plausible that some aerobic bacteria would thrive in an oxygen-rich environment. Recent work involving respiratory culture samples from critically ill adults, and controlled oxygen exposure of mice, found that hyperoxia alters the bacterial community of the lung, selectively enriching it with oxygen-tolerant bacterial taxa, such as Staphylococcus aureus, and conferring a disadvantage to facultative anaerobes such as Pseudomonas aeruginosa).64 Importantly, this study found that alteration of the lung microbiome appeared to mediate lung injury by hyperoxia, as germ-free mice were protected from oxygen-induced lung injury. To our knowledge, the impact of routine supplemental oxygen use on the respiratory microbiome has not been reported as of this writing, although research is ongoing on the influence of supplemental oxygen on the lung microbiome in cystic fibrosis.65
Our study has several limitations. Most importantly, this was a small, cross-sectional pilot study involving the bilateral nares of only 20 individuals, and our findings should be interpreted with caution given our limited statistical power to detect differences by COPD characteristics. Our study population was limited to former smokers with moderate-to-severe COPD who had agreed to participate in a research study on air pollution. Our findings may not be generalizable to current smokers, or those with milder COPD. Furthermore, we were unable to evaluate a more complete spectrum of COPD characteristics, such as a broader range of age and lung function impairment, or the influence of active smoking, on the nasal biome. Our study compared nasal microbial composition between groups of patients with COPD, but did not determine (e.g., by droplet digital PCR) the absolute microbial burden in the nasal cavity, which may also vary by clinical features of COPD. Lastly, unlike shotgun metagenomic sequencing which sequences all given genomic DNA from a sample and generates strain-level taxonomy profiles, 16S rRNA and ITS2 sequencing target specific regions of the rRNA and therefore, produce lower taxonomy resolution (genus-, species-level), which limits the functional profiling. 16S rRNA and ITS2 sequencing can also be biased due to varying PCR amplification frequencies and incomplete reference databases used for sequence analysis.
We can conclude that the taxonomical composition of nasal biome is heterogeneous in COPD patients, and variance in its composition may be explained in part by patient-level clinical and treatment characteristics, including SAMA use, COPD symptom variability, and eosinophil level. Differences in the composition of the airway microbiome may lead to chronic immunologic responses in the airways that might affect COPD disease severity. The reverse is also possible: that those with more advanced COPD may have an altered airway environment that allows certain microbial and fungal species to flourish. Future work, with longitudinal measures of the nasal biome that compares COPD patients to matched healthy controls, and controlled studies evaluating the prospective influence of COPD treatments are needed to verify these findings and determine if such differences are a cause or consequence of COPD features.
Acknowledgments
Author contributions: MBR, LN, WYS, AJS, LC, JER, and MAB planned the quantitative analyses for the study, which were carried out by LC, TMK, and JER. PSL, LN, WYS, and JEK performed protocol planning, quantitative analyses, and interpretation for the quality control project in adult volunteers. MAB interpreted findings and wrote the first manuscript draft. MBR supervised all aspects of the study, including data collection, analysis, interpretation, and manuscript preparation. All authors contributed to the critical revision of the manuscript and approved of the final submitted version.
We would like to thank the study participants for volunteering their time and effort, and the research staff who assisted in this study, especially Wendy Sun and Jennifer Kang.
Declaration of Interest
The authors have no potential conflicts of interest to declare.