Running Head: Quality of Life/Mortality in Severe Alpha-1 Deficiency
Funding support: Funding was provided by CHEST Foundation and AlphaNet.
Date of Acceptance: January 27, 2023 | Publication Online Date: February 2, 2023
Abbreviations: AAT=alpha-1 antitrypsin; AATD=alpha-1 antitrypsin deficiency; CI=confidence interval; COPD=chronic obstructive pulmonary disease; CT=computed tomography; FEV1=forced expiratory volume in 1 second; FEV1 pred%=forced expiratory volume in 1 second percentage predicted; IQR=interquartile range; NE=neutrophil elastase; Pi=protease inhibitor; QOL=quality of life; RCTs=randomized control trials; REF=reference; SGRQ=St George’s Respiratory Questionnaire; SD=standard deviation
Citation: Ellis PR, Holm KE, Choate R, et al. Quality of life and mortality outcomes for augmentation naïve and augmented patients with severe alpha-1 antitrypsin deficiency. Chronic Obstr Pulm Dis. 2023; 10(2): 139-147. doi: http://doi.org/10.15326/jcopdf.2022.0339
Online Supplemental Material: Read Online Supplemental Material (329KB)
Introduction
Alpha-1 antitrypsin deficiency (AATD) is an autosomal codominant disorder defined by a reduced serum level and diminished functionality of alpha-1 antitrypsin (AAT). AAT is a potent inhibitor of serine proteases, which include neutrophil elastase (NE), and acts to regulate their activity within lung tissue as well as other organs. In AATD, the proteolytic activity of NE persists causing accelerated degradation of the elastin component of alveolar walls.1,2 This can lead to early-onset pulmonary emphysema. There are over 100 variants of the AAT gene (SERPINA1) currently known, the most common being the Z allele which is responsible for the majority of severe AATD worldwide with AAT levels typically below the threshold recognized as being susceptible to emphysema development.3
Correction of this protease-antiprotease imbalance is the basic concept behind AAT augmentation therapy, which involves the intravenous administration of AAT purified from human plasma. AAT augmentation therapy was approved in the United States and several other countries in the late 1980s based on its ability to increase human blood and lung levels of AAT. It is currently the only specific disease-modifying treatment for patients with AATD and has been shown to slow emphysema progression as measured by computed tomography (CT) lung density.4-7 Following the results of the RAPID trial,6 which showed a reduction in the rate of CT lung density decline, the European Medicines Agency approved one form of AAT augmentation therapy in Europe. Though CT lung density is now emerging as a reliable surrogate endpoint for emphysema progression,7,8 the efficacy of AAT augmentation therapy has been questioned as randomized control trials (RCTs) have so far failed to show benefits in accepted clinical endpoints such as lung physiology, mortality, and quality of life (QOL).9 Designing an adequately powered RCT that is large enough and long enough to detect disease-modifying effects is not feasible,10 especially since AATD is a rare disease and emphysema, even in AATD, progresses slowly.
Since a large and prolonged RCT approach has not been possible in this condition, it seems logical to explore other experimental designs to evaluate clinically meaningful endpoints. Prospective cohort studies have the potential of studying a greater number of patients with longer follow-up.11-13 We have taken advantage of worldwide variations in access to AAT augmentation therapy. In the United States, augmentation therapy has been administered to patients since 1989 whereas the United Kingdom is yet to approve funding. By comparing mortality and QOL outcomes of these 2 highly characterized patient cohorts, both in Western health care systems and with similar ways of managing other therapies pertinent to AATD lung disease (e.g., inhaled therapies14 and transplantation15,16), we aim to address whether AAT augmentation therapy improves survival and health status in patients with AATD-related lung disease.
Methods
We analyzed QOL outcomes and length of survival in patients with severe AATD-related lung disease in 2 large, prospectively followed cohorts that aimed to characterize the progression and outcomes of severe AATD-related lung disease. Adult patients (≥18 years old) with lung disease and a severely deficient genotype (protease inhibitor [Pi]*ZZ, Znull or Null/Null but excluding Pi*SZ) were included. Written informed consent was obtained from all participants and the study protocol was approved following an independent ethical review in both the United States and the United Kingdom separately. Two primary outcomes were assessed: patient-reported QOL and overall length of survival (all-cause mortality). Figure S1 in the online supplement summarizes patient selection; study size was thus determined by a fixed available number of eligible patients.
Treatment Cohort
The treatment group consisted of individuals in the United States prospectively followed up by AlphaNet, a not-for-profit organization that provides a telephone-based disease management program designed for individuals with AATD-associated lung disease who are prescribed augmentation therapy.17 A weekly dose of 60mg/kg was used for 94.2% of participants, 120mg/kg every 2 weeks for 4.3%, and monthly for 1.5%. Referral to AlphaNet occurs after an individual has been prescribed augmentation therapy by a physician. At present, AlphaNet follows the majority of individuals in the United States who are prescribed augmentation therapy for lung disease due to AATD.
AlphaNet began providing basic disease management in 1999, which consisted of monthly phone calls from trained AlphaNet coordinators. These unstructured phone calls were designed to support patients but did not involve data collection. Since 2008 AlphaNet has used structured telephone interviews to collect extensive information from individuals enrolled in AlphaNet. The current analyses include individuals who completed a telephone interview with AlphaNet between 2008 and the end of 2015. The baseline assessment uses patient self-reports to collect demographic information, AAT genotype (confirmed by physician-reported genotype when possible), year in which the patient began using augmentation therapy, current smoking status and smoking history, and information relating to the prescription and delivery of AAT augmentation therapy. For individuals who reported starting augmentation therapy prior to the year in which they completed their baseline assessment, all analyses in this report use age and smoking status when the individual started using augmentation (rather than age and smoking status when the individual completed the telephone interview). All individuals in the treatment cohort self-reported at baseline that they have lung disease and currently use augmentation therapy. Self-reported outcomes are consistent with the treatment guidelines and insurance requirements for receiving augmentation therapy in the United States. QOL was measured consistently from 2008 onwards using the St George’s Respiratory Questionnaire (SGRQ),18 which measures impact on overall health, daily life, and perceived well-being in patients with respiratory disease. The total score is calculated from 3 domains: symptoms, activity, and impact. Each domain is scored from 0 to 100, with higher scores representing worse QOL. A change of ≥4 SGRQ total points has been found to be a clinically noticeable effect.19 Individuals who are enrolled in AlphaNet complete the SGRQ approximately once per year. The initial data source for mortality was a direct notification from the patient’s family. All individuals reported deceased or that were lost to follow-up as of December 31, 2016, were cross-checked against data provided by the U.S. National Death Index to determine life status.20 Records of patients with missing data were rechecked and excluded if not available.
Control Cohort
The control group consisted of patients with severe AATD (as defined above) who had never received AAT augmentation therapy, from an observational study database of patients in Birmingham, United Kingdom. Annual data collection has been described in detail previously.21 Briefly, participants were referred from either primary or secondary care to a single center from 1996 to 2015. Follow-up included annual medical history and physical examination, post-bronchodilator pulmonary function tests, SGRQ, and baseline AAT genotype. CT thorax was performed by the referring physician or as part of the cohort study to confirm the presence of emphysema. Control participants who had received short periods of augmentation therapy as part of a clinical trial or for other indications (e.g., panniculitis) were excluded from analysis. Mortality was collected from a central National Health Service database or from a direct notification by relatives of the deceased.
Cohort Matching
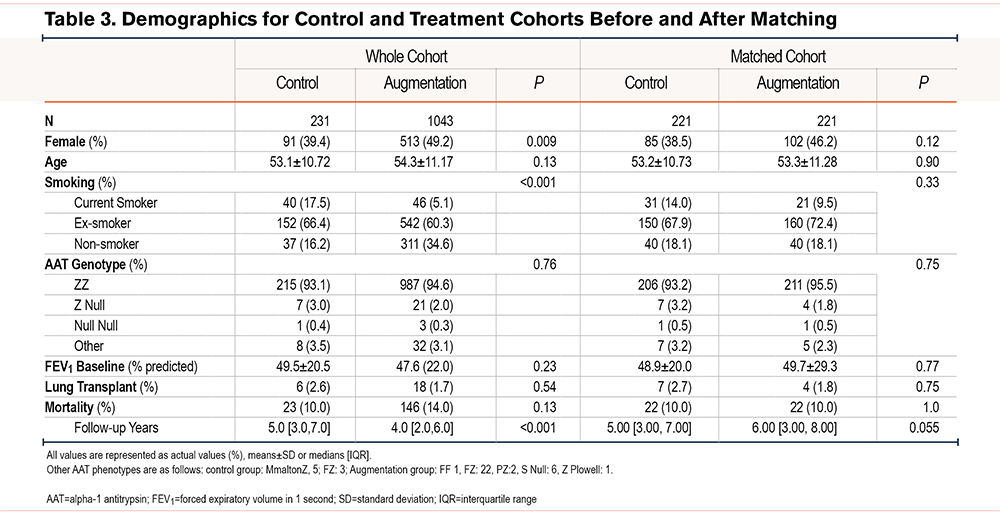
For mortality analyses, one-to-one nearest neighbor propensity matching from the Birmingham AATD Registry with eligible U.S. patients was performed with the matchit package22 in R 3.6.0 using sex, age, patient-reported smoking status (never-smoker, former smoker, or current smoker)23 and baseline forced expiratory volume in 1 second percentage predicted (FEV1 %pred) as matching variables. Former smokers included patients with self-reported cessation of smoking prior to initiation of augmentation therapy or baseline assessment.24 Never smokers included participants who had smoked no more than 20 packs in their lifetime. To reflect improvements in the care of AATD-related lung disease over time in both cohorts,25 a fifth matching variable called “start year” was used, defined as the year of the start of augmentation (treatment) or initial review (control). The balance of covariates was checked and deemed satisfactory prior to statistical analysis. Covariate balance was assessed by comparing absolute mean differences between unmatched and matched cohorts and means using t-tests and Chi-squared tests for normally and non-normally distributed variables.
Statistical Analysis
All statistical analysis was performed using R version 3.6.0 with review by all authors.2 QOL data was used for patients starting augmentation therapy or baseline assessment within 12 months from 2008 through December 31, 2016, since reliable data was available for both cohorts during this time period. Scores recorded after lung volume reduction surgery or lung transplant were excluded from analyses. The dependent variable was defined as change in the SGRQ total score calculated as annual change/year for all patients with 3 or more recorded SGRQ scores. Covariates for the model identified by prior univariate analysis were included if the coefficient p-value was ≤0.1 (Table S1 in the online supplement) or deemed important factors by the authors. The selected covariates were sex, age, first recorded SGRQ total score, and treatment group (augmentation or control). In addition, binary variables for the presence of asthma and bronchiectasis were included to account for differences in disease prevalence between the groups. Multivariable linear regression assumptions were tested and deemed to be achieved including normal distribution of variables, linear relationship between independent and dependent variables, and homoscedasticity.26
The survival package27 was used for all time-to-event analyses using Kaplan-Meier survival curves.28 Survival was calculated from the time of augmentation therapy initiation (augmentation group) or baseline assessment (control group) until time of death. A censor date was set prior to analysis, such that patients alive after December 31, 2016, were censored. In addition, lung transplant recipients were censored in the year of transplantation. Those who were lost to follow-up were censored at the time of withdrawal or the last recorded follow-up visit. All survival times for the augmentation group were reported as integer values (years) in order to comply with the deidentification standards of the U.S. Health Insurance Portability and Accountability Act; control group survival times were transformed to mirror this and thus, reduce potential bias.
The initial protocol for this study set out to assess the survival benefit of augmentation therapy from the earliest time point possible. The implementation of a new questionnaire in 2008 for the augmentation group meant that participants starting augmentation prior to 2008 were subject to immortal time bias. The authors felt it necessary to exclude the pre-2008 participants from the main survival analysis to reduce potential bias.
Results
Quality of Life
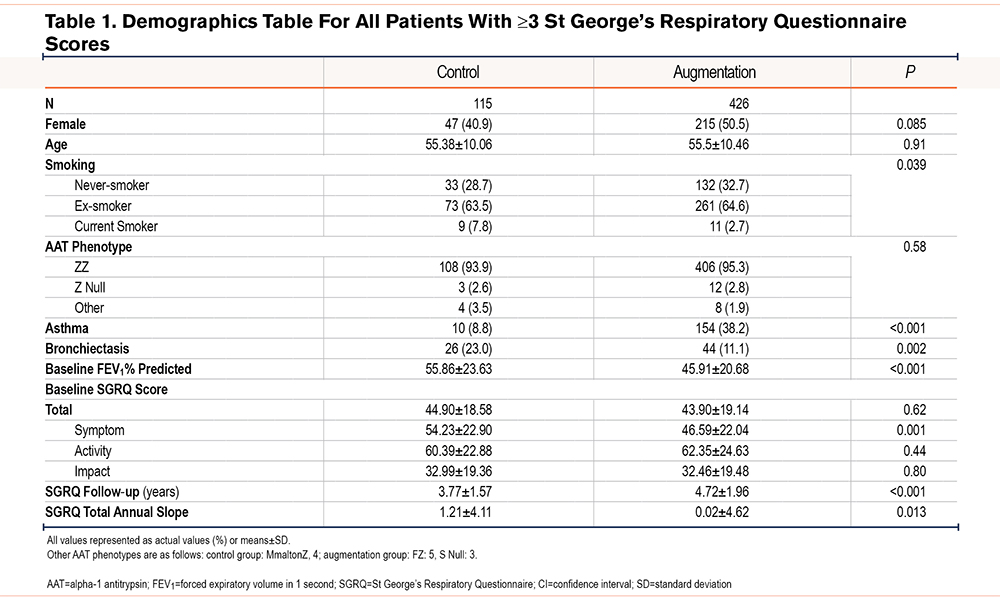
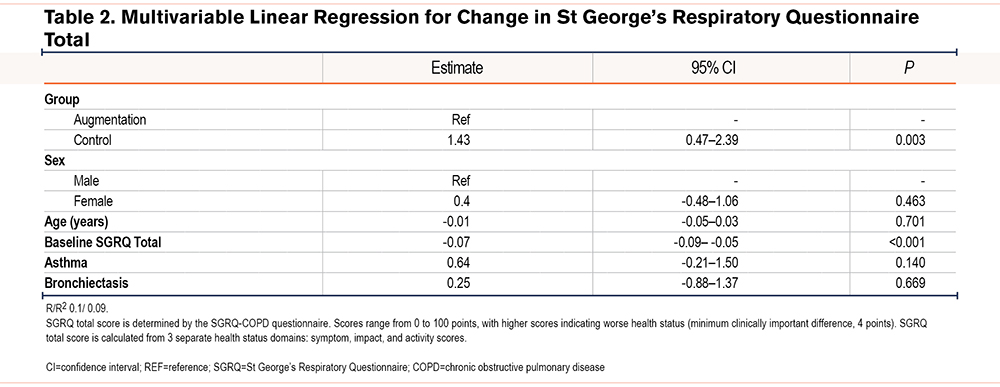
Patient characteristics for QOL analyses are summarized in Table 1. The average baseline SGRQ symptom score was lower in the augmentation group although the total SGRQ score was similar. The mean slope of total SGRQ was 1.19 points/year higher in the control group on average. Results of multivariable linear regression for annual change in total SGRQ score are reported in Table 2; annual decline in health status, reflected as an increase in SGRQ total score, was on average 1.43 points higher/year in the control cohort compared to those receiving augmentation therapy (95% CI 0.47 to 2.39, p=0.003) after adjustment for sex, age, baseline total SGRQ score, asthma, and bronchiectasis. Whilst the slope of total SGRQ was influenced by baseline SGRQ scores the effect was marginal, being only -0.07 points per additional starting SGRQ point (95% CI -0.09 to -0.05, p <0.001). The SGRQ symptom score was higher in the control group at baseline.
Mortality
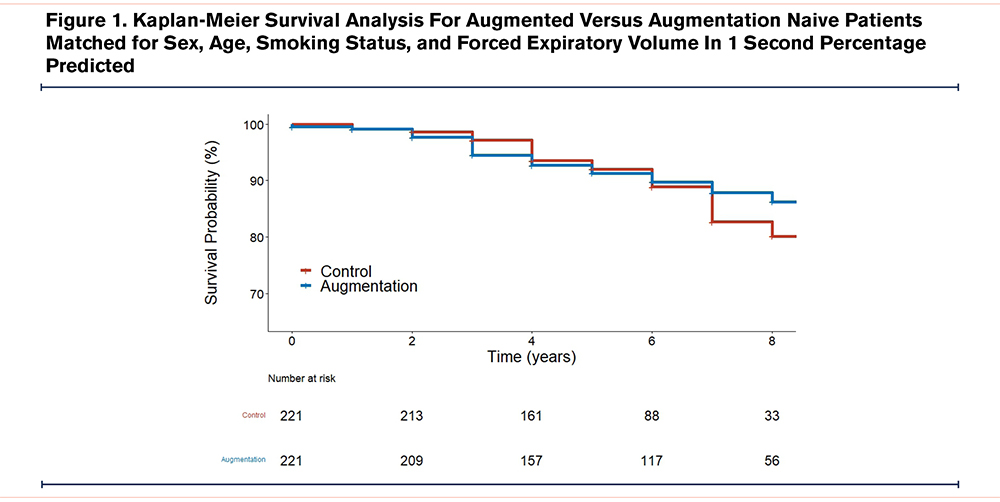
Patient characteristics for mortality analyses pre- and post-matching are summarized in Table 3. Kaplan-Meier survival analysis was conducted to determine whether AAT augmentation therapy affected time to death from all causes following matching (Figure 1). At 7 years, median survival was 82.7% (95% CI 75.3 to 90.7) for the control group versus 87.8% (95% CI 82.8 to 93.2) in the augmentation group, p=0.66.
Discussion
The observational approach of this study attempts to address some limitations associated with RCTs of AAT augmentation therapy which are of insufficient power to assess mortality and QOL. This is largely due to the practicalities of a placebo-controlled study of adequate length and the number of patients required for this rare disease. Two recent RCTs of augmentation therapy versus placebo in severe AATD demonstrate this; both included SGRQ as a secondary outcome and reported no difference between treatment groups.5,6 Neither study was powered to study mortality. The availability of augmentation therapy to patients with severe AATD in multiple centers worldwide makes future enrollment into placebo-controlled RCTs increasingly difficult.
CT lung densitometry has emerged as a promising surrogate marker of emphysema decline7 though its relationship to clinically relevant outcomes remains controversial.9 A systematic review (n=112 studies) demonstrated the relationship of CT lung density to clinically relevant measures, such as FEV1, QOL, exacerbations, and mortality in studies of both usual and AATD-related COPD,29 but the heterogeneity of study designs for CT densitometry made comparisons challenging.
Our study has provided unique data assessing the efficacy of augmentation therapy in patients with lung disease secondary to severe AATD in conventional respiratory outcomes including QOL and mortality.
Quality of Life
Results from this study show an annual decline of SGRQ was on average 1.43 points per year lower for those receiving augmentation therapy. Extrapolation of data from this study calculates benefit of augmentation therapy occurs after 3 years of use, assuming the minimally clinically important difference for SGRQ as ≥4 points.19 Though these results suggest AAT augmentation therapy helps preserve QOL when compared with standard COPD care, there were notable differences between the 2 groups. The control group baseline FEV1% predicted was higher than those receiving AAT augmentation therapy with a higher prevalence of asthma. There was also a higher prevalence of current smokers and individuals with bronchiectasis in the control group. Unmeasured factors, such as exacerbation frequency and other comorbidities, were unaccounted for. These differences may partially explain the results seen. The longitudinal analysis also assumes SGRQ decline is linear. The results should be considered with this in mind.
The anti-inflammatory effects of AAT augmentation therapy are well studied in AATD, in addition to graft versus host disease30 and transplant medicine.31,32 Reduced exacerbation intensity or frequency is one possible mechanism for improvement in QOL with AAT augmentation therapy. The results of the RAPID study did not demonstrate a reduction in annualized exacerbation rate,6 though symptom intensity was not measured directly.
The significance of SGRQ as a marker of disease activity is supported by recently published data in non-AATD COPD from the ECLIPSE study cohort. Celli et al33 found patients with worse SGRQ after 12 and 36 months had higher mortality rates after 8 years. Participants from our study and ECLIPSE were followed up prospectively in a way that reflects a real-world setting that limits bias associated with close patient surveillance that occurs in RCTs. This has been difficult to achieve in AATD-related lung disease10 for the reasons described above and is one of the main strengths of this study.
Mortality
The U.K. registry has shown that CT lung density decline correlates with FEV1 decline34 and mortality in PiZZ AATD, with the strength of the signal being greatest in the upper zone, where progression occurs later in the disease.8 Since augmentation has consistently been shown to reduce emphysema progression demonstrated by CT densitometry, it is predicted to influence long-term mortality.
In this study, over 7 years there was no difference in survival between groups, though overall mortality was low at 10%. This could reflect an earlier diagnosis of AATD-related lung disease and improvements in overall care.35
A recent study by Rahaghi et al reported a survival benefit in U.S. recipients of augmentation therapy versus those never receiving it.36 The participants were not matched and the authors acknowledge several sources of potential bias. These included socioeconomic factors that influence access to medical insurance and physician choices to not prescribe augmentation therapy to those deemed too sick to benefit.
The current study is unique in having reported the first propensity-matched survival data for an extended time between augmentation naïve and augmented patients. Though socioeconomic data was not available for comparison, the risk of bias from this was low since augmentation therapy is covered by Medicare and Medicaid in the United States and, therefore, access to augmentation therapy is not restricted to individuals who are wealthy or who have private insurance. Other comorbidities, including cardiovascular disease and mental health, were not compared and may influence the interpretation of the findings. This study has highlighted the profound challenge in determining the mortality benefit of AAT augmentation therapy.
Strengths and Limitations
The main strength of this study is the large number of patients with AATD-related lung disease, including augmentation naïve and augmented patients, in a real-world setting studied over an extended period of time. This has allowed for the presentation of novel and crucial data in informing the AATD community of the effectiveness of augmentation therapy.
Despite these strengths, the authors are aware of limitations that affect both the quality of life and mortality findings, some of which have been addressed above.
Comparison of 2 different health care infrastructures with limited data on physical and mental health comorbidities meant that the analysis of QOL and mortality outcomes was vulnerable to bias. Studies have indicated that telephone or face-to-face administration of SGRQ are equally valid37 and mortality is less likely to have been affected by differing patient data collection as mortality is a finite outcome confirmed in registry data.
It is unclear whether SGRQ is prone to psychosomatic bias in the context presented. Both groups have been informed they have a rare disease that is potentially fatal though only the augmentation group has access to the best (and only) disease-modifying therapy available. The control group is denied access as a result of a lack of access to augmentation therapy in the United Kingdom which may negatively impact their scores. These factors should be taken into consideration when interpreting the QOL and mortality results.
Conclusion
Comparison of 2 highly characterized longitudinal AATD cohorts was not able to reliably confirm if AAT augmentation therapy improves QOL or mortality in patients with severe AATD- related lung disease. Unless large-scale studies, such as genetic testing at birth, are implemented, it is unlikely that the effect of augmentation therapy on mortality versus placebo will be determined in a meaningful analysis. In a rare disease, such as severe AATD-related lung disease, it may be pragmatic to measure treatment response from augmentation therapy via more attainable clinical endpoints, such as CT density, to inform treatment decisions in AATD- related lung disease.
Acknowledgments
Author contributions: PE, RASa, RASt, and AT contributed to the concept of the study. Data acquisition was supported by PE, RC, and KH. Data analysis was performed by PE with data interpretation agreed upon by all authors. PE wrote the initial manuscript with review by all authors prior to submission.
Declaration of Interest
PE and RC have no conflicts of interest. KM reports personal fees from AlphaNet. DM reports salary and shares of stock from GlaxoSmithKline, outside the submitted work. RASt reports grants and personal fees from CSL Behring, and personal fees from Boehringer, Astra Zeneca, Mereobiopharma, Shire, Chiesi, and Akari, outside the submitted work. RASa is employed by AlphaNet, reports personal fees from Grifols and CSL Behring, non-financial support from Inhibrx and Arrowhead, and grants from Matrx, outside the submitted work. AT reports grants from Grifols biotherapeutics and the Alpha-1 Foundation, personal fees from CSL Behring, and grants and non-financial support from Arrowhead, outside the submitted work.