Running Head: Augmentation Therapy and Systemic Inflammation
Funding Support: This work was supported by the Alpha-1 Foundation.
Date of Acceptance: June 11, 2023 │ Published Online Date: June 21, 2023
Abbreviations: AAT=alpha-1 antitrypsin; AATD=alpha-1 antitrypsin deficiency; Aug=augmentation therapy; COPD=chronic obstructive pulmonary disease; CRP=C-reactive protein; ER=endoplasmic reticulum; FEV1=forced expiratory volume in 1 second; IL=interleukin; %pred=percentage predicted; PI=protease inhibitor
Citation: Lascano J, Riley L, Khodayari N, Brantly M. Augmentation therapy modulates systemic inflammation in individuals with alpha-1 antitrypsin deficiency and chronic obstructive pulmonary disease. Chronic Obstr Pulm Dis. 2023; 10(3): 308-316. doi: http://doi.org/10.15326/jcopdf.2023.0407
Introduction
Alpha-1 antitrypsin (AAT), an acute-phase protein, is the most abundant serine protease inhibitor (PI) in the plasma.1,2 This glycoprotein is mainly synthesized in the liver by hepatocytes and secreted into circulation to maintain plasma levels of 20 to 53µM.3,4 The SERPINA1 gene coding AAT is located on chromosome 14, and the majority of people carry 2 normal genes, designated as the M variant.1
Alpha-1 antitrypsin deficiency (AATD) is an autosomal codominant genetic disorder characterized by a low level of circulating AAT. The most clinically important allelic variants in individuals with AATD include the Z variant (Glu-342-Lys), S variant (Glu-264-Lys), and null variants. The Z variant is the most common variant associated with clinical manifestations of the disease. Individuals with the Z variant of AAT produce a mutated protein that folds abnormally, causing polymerization and accumulation of misfolded AAT in the endoplasmic reticulum (ER) in the hepatocytes and other cells that produce AAT. The ER stress mediated by the accumulation of misfolded AAT causes cellular damage by a gain of toxic function mechanism.5 Moreover, the accumulation of misfolded AAT within the hepatocytes results in a decreased level of AAT in the systemic circulation. Low levels of AAT in the systemic circulation leads to the unopposed activity of neutrophil elastase, which is the major target for the protease inhibitory function of AAT. Profound activity of neutrophil elastase, a serine protease produced by activated neutrophils in the lungs, results in significant lung alveolar damage with a progressive decrease in forced expiratory volume in 1 second (FEV1) associated with the development of emphysema.6 Although the main role of AAT is inhibition of neutrophil elastase activity to protect the lung matrix from degradation, AATD is mostly considered as an inflammatory disorder, wherein the neutrophils play a critical role in various inflammatory processes in different tissues.7
Chronic obstructive pulmonary disease (COPD) is a progressive pulmonary disease associated with systemic inflammation.8 Increased levels of several systemic inflammatory markers including C-reactive protein (CRP) are associated with the progression of inflammatory diseases such as COPD and AATD.4,9 However, AATD individuals with COPD do not experience the same inflammatory response as non-AATD COPD patients.10 Individuals with AATD can present at a younger age and have a higher risk of progressive decline of FEV1 and radiologic evidence of emphysema. Risk factors for the development of COPD in individuals with AATD include smoking, male sex, asthma, and chronic bronchitis.2 Currently, the only specific FDA-approved treatment for AATD individuals with COPD is a weekly infusion of purified AAT, termed augmentation therapy. The criteria for treatment include moderate to severe COPD as per the Global Initiative for Chronic Obstructive Lung Disease criteria5 and a low level of AAT (<11µM/dL).11 AAT augmentation therapy has shown efficacy in AATD individuals with COPD by maintaining circulating levels of AAT and effective inhibition of neutrophil elastase in the lung.12 In this study, we examined the effect of AAT augmentation therapy on the systemic inflammation in individuals with COPD due to AATD using plasma levels of CRP as our main outcome.
Study Design and Methods
Study Participants
Study participants with ZZ genotype were chosen from the Alpha-1 Foundation DNA and Tissue Bank project at the University of Florida (IRB#201500842). Age and sex matched never-smoking participants with a MM genotype and no family history of AATD were selected from the Alpha-1 Foundation DNA and Tissue Bank for use as a control group. The Alpha-1 Foundation DNA and Tissue Bank collects medical information and human tissue of individuals with AATD from across the United States. The Alpha-1 Foundation DNA and Tissue Bank project is sponsored by the Alpha-1 Foundation and is physically located at the University of Florida, College of Medicine. The University of Florida Institutional Review Board approved the Alpha-1 Foundation DNA and Tissue Bank protocol and analysis of the data presented in this study.
Upon enrollment, individuals signed informed consent and completed a registration form and extensive medical questionnaire. Smoking history was reviewed, and 40% of participants with AATD were previous smokers and 3% were current smokers at the time of enrollment. All control participants were never smokers. All pulmonary function measurements were recorded at the time of the initial questionnaire, and FEV1 values were measured in liters. For the purpose of the study, participants with a proven PI*ZZ genotype (by Taqman allelic discrimination) were included. AAT phenotype was determined using an isoelectric focusing assay.
AATD individuals were divided into 2 groups: (1) augmented (patients receiving AAT augmentation therapy at the time of recruitment in the study), and (2) non-augmented (patients not receiving AAT augmentation therapy at the time of recruitment in the study). The treatment decision for the AATD individuals with AAT augmentation therapy was made by each respective individual’s physician. The Alpha-1 Foundation DNA and Tissue Bank did not intervene in any way in the treatment plan of individuals participating in this study.
A secondary analysis was performed by investigating the effect of AAT augmentation therapy on systemic inflammation in participants with an FEV1 above and below 50% predicted to better understand this effect regardless of lung function.13
Plasma Levels of Alpha-1 Antitrypsin and C-Reactive Protein Quantification
Serum levels of AAT and CRP were obtained using a Dade Behring Nephelometry as previously described by our group.11 Briefly, plasma samples were centrifuged at 20,000 x g for 10 min to remove fibrin or debris. Samples (100μL) were then loaded undiluted, and AAT and CRP levels were measured on a Siemens Nephelometer calibrated with the Rheumatology Standard according to the manufacturer’s directions. The assay has a range of 0.1–50mg/L.
Statistical Analysis
Levels of CRP were compared between controls and AATD participants as well as between augmented and non-augmented AATD individuals. Subgroup analyses were performed in each group between those who had an FEV1 value greater or less than 50% predicted. All comparisons were determined using an unpaired t-test and standard Pearson’s correlation. Data are expressed as mean±standard deviation. A P-value less than 0.05 was considered significant. All statistical analyses were performed using PRISM software, version 8.31 (San Diego, California).
Results
Baseline Characteristics of the Study Population
A total of 742 AATD individuals with PI*ZZ genotype and 40 control participants were tested for plasma levels of CRP. Of those, 498 participants (67.1%) were on AAT augmentation therapy and 244 participants (32.9%) were not on augmentation therapy. The mean age of the participants having AAT augmentation therapy was 63.5±9.6 years, which was higher when compared to the participants who were not AAT augmented (57.9±14.9 years) (P<0.0001). The percentage of males and females was similar in both groups of paricipants. The plasma levels of AAT were lower in the non-augmented group (4.64±1.59µM) compared with the AAT augmented group of participants (20.0±10.7µM), (P<0.0001) as expected. We observed that the circulating levels of CRP were elevated in AATD individuals regardless of AAT augmentation status, compared to healthy controls (0.18mg/dL versus 0.06mg/dL; P<0.0001) (Figure 1). Similarly, there was a significant difference in the plasma levels of CRP in the AAT augmented group of participants versus the non-augmented group of participants (0.36±0.79mg/dL versus 0.56±1.65mg/dL; P=0.0245) (Table 1). Finally, when CRP levels of controls were compared to augmented participants, there was still a significant difference (0.06mg/dL versus 0.36mg/dL; P<0.001).
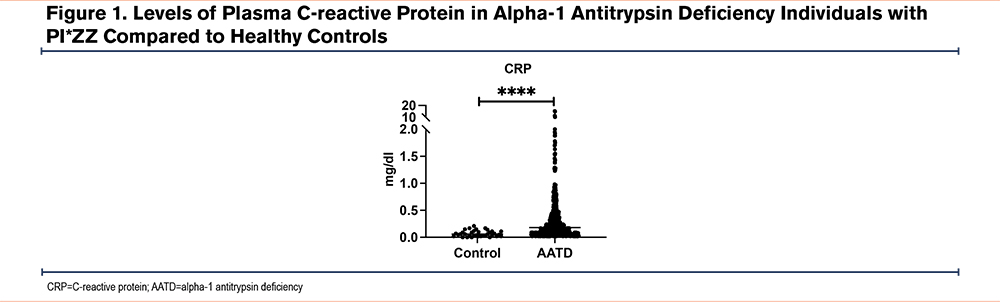
Correlations Among Plasma C-Reactive Protein and Lung Function in Alpha-1 Antitrypsin Deficiency Individuals
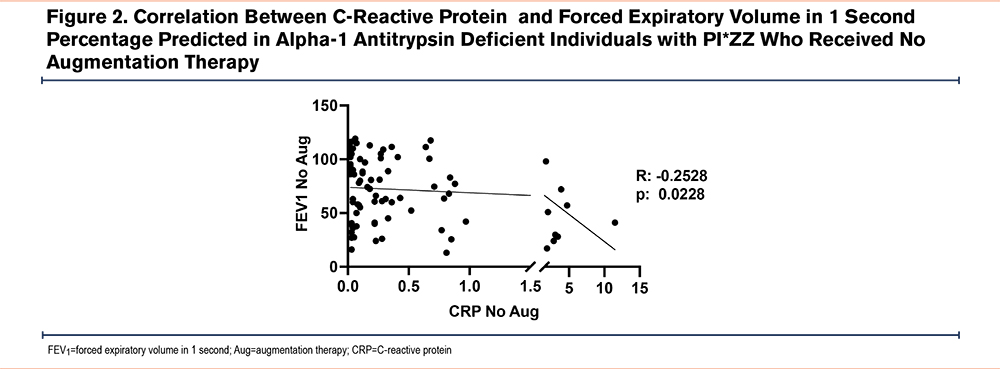
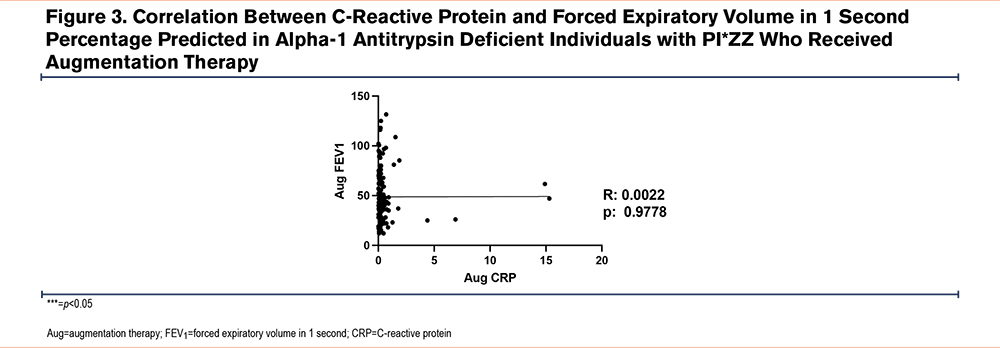
Next, pulmonary function test data from 508 patients were analyzed. Of that subset, 342 (67.3%) patients were in the AAT augmented group and 166 (32.7%) never received AAT augmentation therapy. We found that although the AAT augmented group had worse lung function as compared to AATD individuals without AAT augmentation therapy (46.4±23.2 FEV1 percentage predicted [%pred] versus 73.9±29.7 FEV1 %pred; P<0.0001) (Table 2), they had significantly lower levels of plasma CRP (0.32±0.54µM versus 0.69±1.97µM; P<0.05). Furthermore, we examined the correlation between CRP levels and FEV1 in the group of AATD individuals not receiving AAT augmentation and found a mild negative correlation (r=−0.2528, P<0.05) (Figure 2). In contrast, no similar correlation was observed in the participants receiving AAT augmentation therapy (Figure 3).
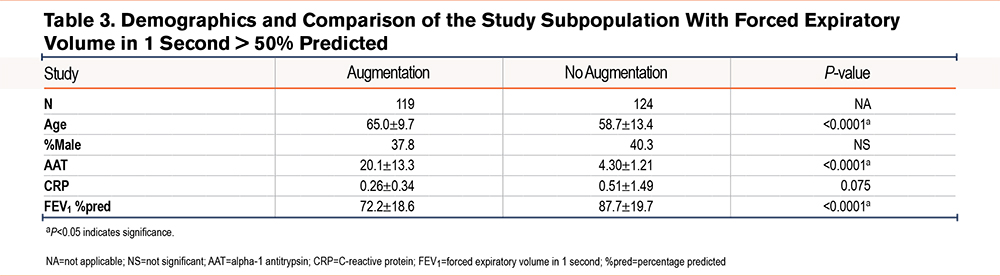
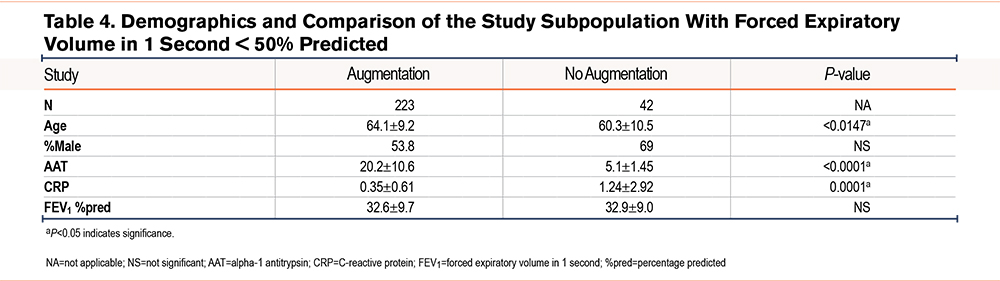
We further divided our study population based on FEV1 values greater than 50% of predicted (243 patients; 47.8%) and FEV1 values less than 50% of predicted (265 patients; 52.2%). Both groups maintained similar demographic comparisons in terms of age, sex, and AAT levels as the entire cohort. We observed no significant differences between the levels of CRP (0.26±0.34µM versus 0.51±1.49µM; P=0.075) in augmented and non-augmented patients despite having a significant difference in FEV1 %pred in AATD patients with FEV1 values greater than 50% of predicted (72.2±18.6% versus 87.7±19.7%; P<0.0001) (Table 3). However, 223 patients with FEV1 values less than 50% of predicted (84%) with AAT augmentation therapy had significantly lower CRP levels compared with 42 patients (16%) who didn’t receive AAT augmentation therapy (1.24±2.92mg/dL versus 0.35±0.61mg/dL; P<0.0001) (Table 4), despite having a similar FEV1 %pred (32.9±9.0% versus 32.6±9.7%; P>0.05) respectively.
Discussion
In our study and consistent with previous studies,14 we showed elevated levels of CRP in AATD individuals as compared to control participants, indicating an ongoing systemic inflammation in the participants with AAT deficiency. AAT is an acute-phase protein with anti-inflammatory properties. Therefore, the lack of anti-inflammatory properties of AAT plays the major role in AATD-mediated COPD.15 It is well-recognized that COPD patients have significant signs of systemic inflammation.16 It has been also shown that plasma levels of CRP are elevated in COPD patients when compared with nonsmokers or smokers without airway obstruction.17 Furthermore, it has been shown that the severity of COPD manifestations is associated with increased levels of CRP, reflecting the systemic burden of inflammation.18 In the presented study, we examined the plasma levels of CRP in AATD patients with COPD, regardless of the AAT augmentation therapy status, and show that individuals with AATD-mediated COPD have higher plasma levels of CRP than those non-AATD-COPD individuals, which has been previously reported in the literature.19 Our study adds to these existing data by emphasizing the role of AAT in harnessing systemic inflammation, in addition to lung tissue specifically.
Consistent with our previous report,14 our AATD study participants had significantly higher plasma levels of CRP compared to health controls, indicating mild levels of systemic inflammation. When we compared the level of circulating CRP in AATD patients with COPD between those receiving and not receiving augmentation therapy, we observed an inverse correlation between the CRP levels and FEV1 in AATD individuals without AAT augmentation therapy. However, this correlation was not observed in AATD individuals who received AAT augmentation therapy, as most of those individuals’ CRP levels were lower and closer to the normal range, despite being a much older population with greater airflow limitation as reflected in their FEV1 values. Previous population-based studies in COPD patients have clearly shown a relationship between higher CRP levels and worsening of airflow obstruction and FEV1 decline.20,21 AATD patients in our AAT augmented group had moderate to very severe obstruction; notably, not only did they significantly have lower levels of CRP when compared to the higher FEV1 non-augmented group, but they also lacked the inverse correlation between CRP levels and FEV1. The absence of a correlation in the AAT augmented cohort may be due to the lower CRP levels, and that they were congregated around zero, thereby, making it difficult to establish a correlation between CRP levels and FEV1. Another possible explanation for this observation would be a change in the course of their disease, especially in terms of FEV1 decline due to augmentation therapy itself.
Next, we expanded our study by dividing our study population based on FEV1 values greater or less than 50% of predicted. This separation within our cohort allowed us to confirm the role of augmentation in lowering the CRP levels in this subgroup. We found significantly lower CRP levels in the augmented group compared with the non-augmented group, despite both groups having very similar FEV1 ranges. This finding adds to the body of evidence that augmentation therapy might be beneficial regardless of FEV1. Dahl et al showed that a CRP level greater than 3mg/L is associated with a higher risk of hospitalizations and mortality in COPD patients when compared with CRP levels less than 3mg/L.22 Among the patients with less than 50% predicted FEV1 in our study, the CRP levels in the non-augmented group (1.24±2.92mg/dL) were higher than in the augmented group (0.35±0.61mg/dL), confirming the finding that augmentation therapy reduces CRP levels in patients with AATD and severe COPD.
The results from the presented study demonstrate the effects of AAT augmentation therapy on systemic inflammation in patients with AATD and severe COPD. Despite the significant advances in our understanding of the mechanisms of this disease, there is still a very limited understanding of the systemic effects of AAT. Data from in vitro studies have shown that AAT may have non-canonical properties other than neutrophil elastase inhibition. These include interactions with the proteolytic cascade of enzymes involved in apoptosis,23 particularly caspase-, inhibition of superoxide production by neutrophils,24 and reduction of tumor necrosis factor-α lethality.25 AAT modulates lipopolysaccharide-induced inflammation26 and regulates the response of macrophages to pro-inflammatory stimuli.27 Lack of these effects might also play a role in the development of disease and outcomes in this population.
Several observational studies, including the largest registry from the National Institutes of Health, have found a decrease in 5-year mortality in the patients receiving AAT augmentation therapy.28 Moreover, Tonelli et al13 reported an improvement in lung function in the participants with AATD and COPD receiving augmentation therapy, when adjusted by age, gender, smoking status, and baseline FEV1. Together with our findings, this data opens the possibility of further exploration of the effects of AAT augmentation therapy in reducing systemic inflammation in COPD populations with or without AATD.
Our understanding of the role of inflammation in the development of COPD has significantly expanded in the past few years. Currently, there is a general consensus in the field that we agree that systemic inflammation negatively impacts the outcome and mortality of COPD. However, this relationship between local and systemic inflammation is more complex and requires further clarification. So far, previous studies have failed to find an association between local and systemic inflammation and demonstrated a difference in the regulation of inflammation in the pulmonary and systemic circulation. For example, there is a lack of association between sputum and plasma concentrations of interleukin-6 (IL-6) and soluble tumor necrosis factor-α receptors in patients with COPD.29,30 All together, these findings suggest that systemic inflammation plays an independent role in the pathophysiology of COPD and that controlling local inflammation might not be sufficient as a treatment option for COPD. Yet, therapeutic approaches continue to focus on bronchodilators and control of local inflammation.31
It is still unclear if reducing the systemic inflammatory burden in COPD patients will have a significant effect on outcomes.32 The effect of inhaled steroids on inflammation is also controversial. Sin et al reported in 2 different trials that the use of inhaled corticosteroids, such as fluticasone, significantly reduced CRP levels in COPD patients with mild to moderate disease.33 They later reported that neither fluticasone alone nor in combination with salmeterol had any effect on CRP or IL-6 levels. They suggested that inhaled corticosteroids might reduce local inflammation but had no effect on the markers of systemic inflammation such as CRP.34,35 These findings leave an open door to continue investigating ways to target systemic inflammation in this population and what effects therapies may have on patient outcomes.
Our study is limited by the fact that it is an observational study. There is also a possibility of selection bias because only individuals with all the data of interest were selected. In terms of the data obtained, spirometry was reported from different centers, and biomarkers were obtained at a single point in time. The decision of augmentation therapy treatment was entirely up to the physicians taking care of the patients. Several reasons can limit the decision to prescribe augmentation therapy, including active smoking, medication compliance, or the perception of good lung function. Despite these limitations, our study shows that augmentation therapy may be able to modulate systemic inflammation presented in the form of CRP in all patients receiving it, but especially in those with a lower than 50% predicted FEV1.
Conclusion
In conclusion, this study demonstrates that the plasma levels of CRP are increased in our study population of AATD individuals with COPD compared to healthy controls. Furthermore, we demonstrated that AAT augmentation therapy in AATD individuals with COPD results in a reduction of CRP levels and more clearly in those with an FEV1 less than 50% of predicted. Our presented data suggest that targeting systemic inflammation may alter the course of the disease in terms of lung function. Finally, we believe that there is a requirement for further clarification of the effects of reducing systemic inflammation in patients with COPD with or without AATD on the comorbidities and outcomes.
Acknowledgements
Author contributions: MB conceptualized and supervised the project, designed the methodology, recruited patients, collected samples, and contributed to the draft manuscript. JL performed experiments, analyzed data, and wrote the original manuscript. NK and LR revised the manuscript and data analysis. All authors read and approved the final manuscript.
Data sharing: Normal and AATD individuals were recruited at the National Institutes of Health Clinical Center after providing consent. The datasets and analysis of this study are available from the corresponding author upon reasonable request.
Declaration of Interest
The authors declare that the research was conducted in the absence of commercial or financial relationships that could be construed as competing interests.