Running Head: Smoking, CMV Serostatus, and Cell Phenotypes in Smokers
Funding support: The project described was institutionally supported and in part by the National Center for Advancing Translational Sciences of the National Institutes of Health, under Award Number UL1TR001425 and HL1159538 and the Veterans Administration (I01BX002347 and I01CX001891). The content is solely the responsibility of the authors and does not necessarily represent the official views of the Funders.
Date of Acceptance: May 23, 2023 │ Published Online Date: June 2, 2023
Abbreviations: AECOPD=acute exacerbation of COPD; BMI=body mass index; CI=confidence interval; CMV=cytomegalovirus; COPD=chronic obstructive pulmonary disease; FEV1=forced expiratory volume in 1 second; FVC=forced vital capacity; IL=interleukin; NK=natural killer cells; NKGC2=natural killer group C2; REA=recombinant antibody
Citation: Burkes RM, Bailey E, Hwalek T, et al. Associations of smoking, cytomegalovirus serostatus, and natural killer cell phenotypes in smokers with and at risk for COPD. Chronic Obstr Pulm Dis. 2023; 10(3): 286-296. doi: http://doi.org/10.15326/jcopdf.2022.0382
Introduction
Chronic obstructive pulmonary disease (COPD) is a leading cause of morbidity and mortality worldwide.1 An important future direction in COPD care lies in the identification of distinct biological mechanisms of disease, known as endotypes,2-4 which can facilitate the development of precision medicine approaches. Among the reported endotypes are high peripheral eosinophilia,5 deranged mucous production and viscosity,6 hypersecretion of interleukin7 (IL)-17, and chronic airway infection.3,8 As of this writing, high peripheral eosinophilia has led to a change in COPD treatment paradigms, while other endotypes remain targets of ongoing research.1,3
Innate lymphocytes, specifically natural killer (NK) cells, show well-documented population differences in humans when accounting for age, chronic cytomegalovirus (CMV) infection, and smoking.9-12 NK cell populations in smokers are found to be fundamentally different from non-smokers12 with NK cells from smokers displaying more airways cell-directed cytotoxicity in vitro.13 Further, background literature suggests that high CMV titers are associated with higher mortality from COPD.14 Latent CMV infection is also thought to mold NK cell populations.15 It is reasonable to speculate that the interface of smoking, CMV, age, and NK cell populations may represent a potential pathway to COPD development and progression. However, relationships between these factors in the COPD population need further investigation. As NK cell activity may be modulated by current16,17 and emerging18 COPD therapies, gaining foundational knowledge of the associations between CMV serostatus and NK cell populations in COPD and how this relationship may be influenced by COPD risk factors is an important first step in the aspirational goal of COPD therapies targeting NK cell biology.
This study investigates a cohort of current and former smokers with and at risk for COPD and reports the independent influence of CMV serology on NK cell populations, identifies factors that may predict NK cell populational differences, and describes the difference in the NK cell-CMV seropositivity relationship in COPD and those with prominent risk factors. Our cohort of 170 Veterans contains well-defined demographic and clinical data and available biosamples to assess the relationship between CMV seropositivity and NK cell populations. We hypothesized positive CMV serostatus will be associated with differential NK cell populations and utilized predictive modeling to describe factors significantly associated with specific NK cell populations. More importantly, we hypothesize that the relationship between positive CMV serostatus and prevalent NK cell populations that emerge due to frequent noxious exposures12,19 will be larger in those with a more extensive smoking history and clinical COPD. The findings of this study will provide foundational evidence for further mapping NK cell populations over time in COPD, determining accrued inflammatory potential, and understanding how population shifts may longitudinally influence clinical COPD outcomes.
Methods
Study Participants
The cohort for this study is made up of 170 participants recruited between 2016 and 2019 at the Cincinnati VA Medical Center. This cohort includes current, former, and never smokers with or at risk for COPD with available whole blood for sampling cellular phenotypes and mediators, and accompanying demographic, clinical, and spirometric data. Participants did not have active cancer or autoimmune disease. Study procedures were performed after obtaining written informed consent from patients. The study design was reviewed and approved by the VA Research and Development Committee and the University of Cincinnati Institutional Review Board (IRB# 2014-2354). All methods were performed in accordance with the relevant guidelines and regulations. Data from patients, such as questionnaires, spirometry results, and all specimens were assigned a unique study number to de-identify patient personal information.
Data Collection
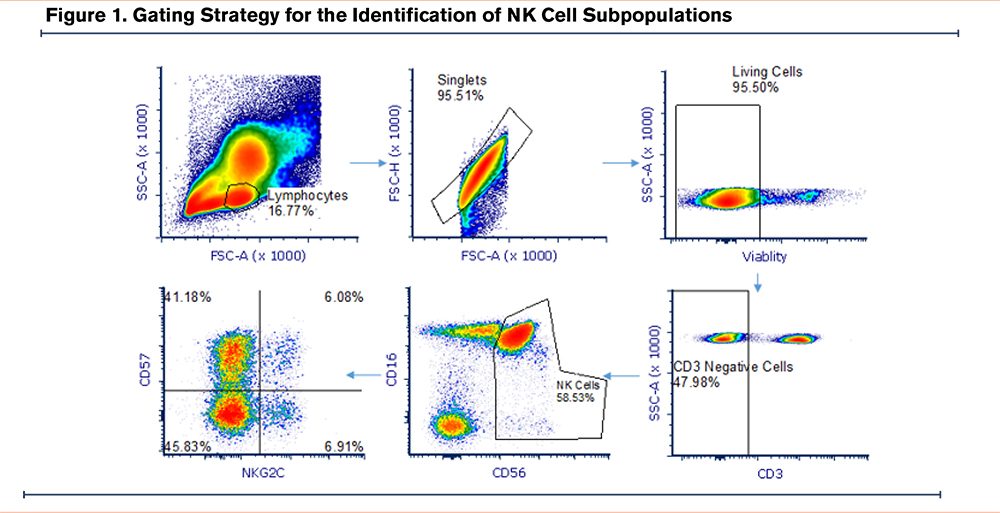
Demographic data (age and reported sex) were collected at enrollment. Smoking status was collected at enrollment and participants were defined as current and former smokers based on the use of combustible tobacco products at the time of enrollment. Pulmonary function testing was performed by American Thoracic Society/European Respiratory Society guidelines20 and absolute and percentage predicted values are reported. The presence of COPD (defined as having a forced expiratory volume in 1 second [FEV1] to forced vital capacity [FVC] ratio <0.70) and disease severity were determined based on Global initiative for chronic Obstructive Lung Disease (GOLD)1 criteria. Participants provided 30 ml of whole blood at the time of enrollment for assessment of NK cell activity as well as testing for CMV serostatus. Multi-parameter flow cytometry was used to determine the %-population of CD57+ NK cells, natural killer group 2C (NKG2C)+ NK cells, and CD57+NKG2C+ NK cells among the total NK cell population with the following antibodies: PE NKG2C (Clone; recombinant antibody 205, Milllitenyi Biotec), APC-CD57 (Clone; human natural killer 1 cells, Biolegend), PECy7-CD56 (Clone; 5.1H11, Biolegend), kiravia blue CD3 (Clone UCHT1, Biolegend), PerCP Cy5.5-CD16 (Clone 3G8, Biolegend), zombie NIR (Viability, Biolegend). Gating strategy depicted in Figure 1.
Assays to assess the presence and antigen-binding avidity of CMV antibodies were performed by ELISA according to the manufacturer’s protocol (VIDITEST anti-CMV IgG and IgG avidity, Vidia, Czechoslovakia ) as previously reported.21 The avidity of antigen binding is presented as high (representing long-standing or repeat CMV exposure), intermediate, and low (representing a recent CMV infection).
Study Design
Descriptive statistics were used to examine differences in demographic and clinical factors among each NK cell subpopulation individually. Pearson’s pairwise correlation was performed to describe relationships between proportions of NK cell populations and continuous variables while Student’s t-test was used to determine the difference in means between categorical values and distribution of NK cell subpopulations.
Bivariable and multivariable linear regression was used to determine the relationship between cytotoxic NK populations, CMV serostatus, and clinically relevant risk fators.22 We further present a predictive modeling approach with the same set of variables that were selected for the linear models based on biological relevance. We then removed variables based on the least strong, least significant association to come to a final, parsimonious model describing the magnitude of association between predictors of cytotoxic NK cell populations and predictive covariables. We describe both the original model and the final model in the results. For CD57+, NKG2C+, and CD57+NKG2C+ populations, original models included positive CMV serostatus, age (in years), Black race, female gender, pack-years smoked, body mass index (BMI), and post-bronchodilator FEV1 %-predicted. As stated above, in the predictive modeling approach, variables were removed in a stepwise fashion to produce a final, parsimonious model.
We performed specific investigation of the effect of CMV seropositivity on NK cell populations stratifying by variables of interest. We separated cohorts by COPD versus not, ≥40 pack-years smoking versus <40 pack-years smoking, and those with ≥40 pack-years smoking and COPD. Bivariable and multivariable linear regression modeling was used to determine the associations between CD57+, NKG2C+, and CD57+NKG2C+ proportions and CMV serostatus. Covariables included age (in years), Black race, female gender, pack-years smoked, post-bronchodilator FEV1, positive CMV serostatus, and BMI. The stratified results allow for numerical comparison between strata. To test for significant influence of the stratifying variables, in a model using entire cohort and the same covariables, we introduced an interaction term of the stratifying variable and CMV serostatus. A result that is statistically significant suggests that CMV serostatus significantly modifies the statistical relationship between the stratifying variable and NK cell receptor populations which we provide as a corollary to illustrate statistical difference between the stratified groups.
The results of linear regression modeling report point estimates with 95% confidence intervals. For all analyses, p<0.05 is considered significant. Missingness in the cohort is infrequent and thought to occur at random. Statistical analyses were performed using STATA version 15.1 (College Station, Texas).
Results
The median age of the cohort was 61 years old (25%–75%, 56–68 years old). Of the cohort, 45% were Black, 6% were female, and 78% were current smokers. The average pack-years smoked was 39 (quartile 1–quartile 3, 13–56 pack years). For lung function, 42% met criteria for COPD and the average post-bronchodilator FEV1 was 2.56 L (quartile 1–quartile 3 1.80–3.10 L). A COPD exacerbation event in the 2 years prior to study enrollment was reported by 33% of participants. Those with COPD and a ≥40 pack-year smoking history comprised 29% of the cohort.
In this cohort, the median proportion of NK cells (identified asCD3+CD56+) expressing CD57 cells was 59.1% (quartile 1–quartile 3, 46.5% –67.9%) (Figure 1), median proportion expressing NKG2C was 8.8% (quartile 1–quartile 3, 4.1%–16.3%), and median proportion expressing both was 4.8% (quartile 1–quartile, 3 1.9%–9.8%).
CD57+ NK Cells
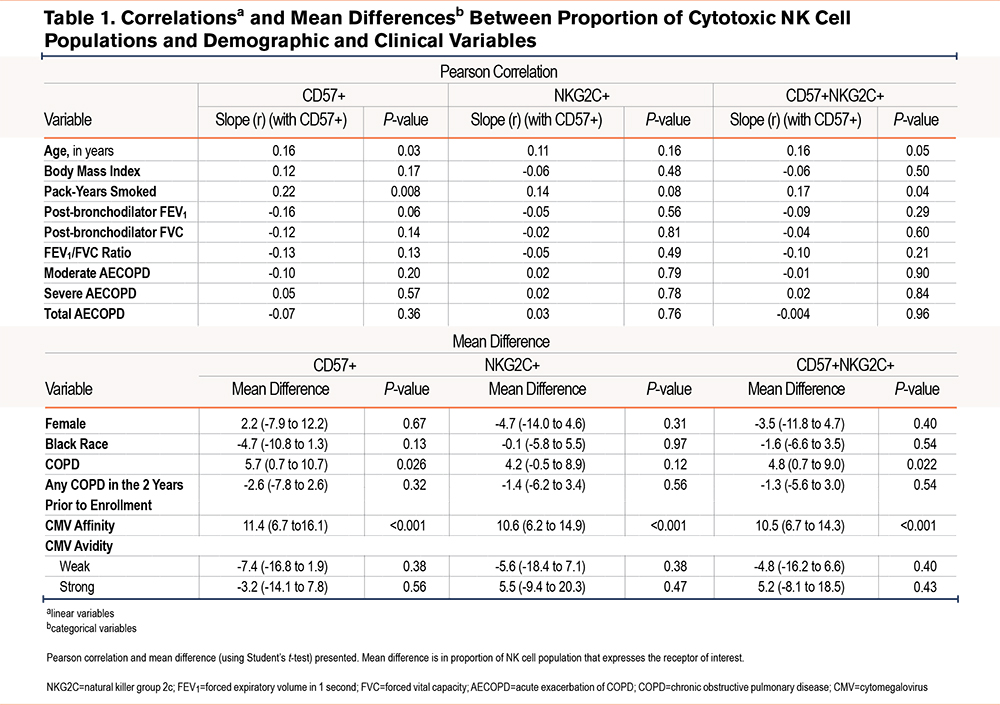
In bivariable analysis, when compared with continuous variables, the %-population of CD57+ NK cells were positively associated with only age (r=0.16). Those with COPD had a 5.7% higher mean difference in %-population CD57+ NK cells (P=0.026) while positive CMV serostatus was associated with an 11.4% higher mean %-population difference when compared to those who were not CMV seropositive (P<0.001) (Table 1).
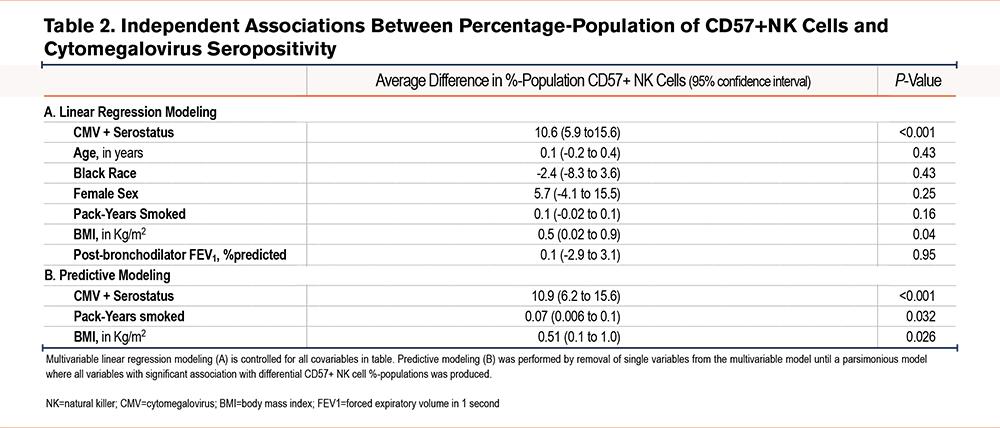
When controlled for clinically relevant covariables, those with positive CMV serostatus had a 10.6 higher %-population of CD57+ expressing NK cells (95% confidence interval [CI] 5.9 to 15.6; P<0.001) (Table 2). From this main model, we removed variables based on lack of association strength and found that positive CMV serostatus (10.9 %-difference in CD57% NK cells [95% CI 6.2 to 15.6]; P<0.0010), pack-years smoked (0.07 %-difference in CD57+ cells per pack year [95% CI 0.006 to 0.1]; P=0.032), and BMI (0.51 %-difference in CD57+ cells per 1kg/m2 increase in BMI [95% CI 0.1 to 1.0]; P=0.026) are all independently correlated with a greater proportion of CD57+ NK cells.
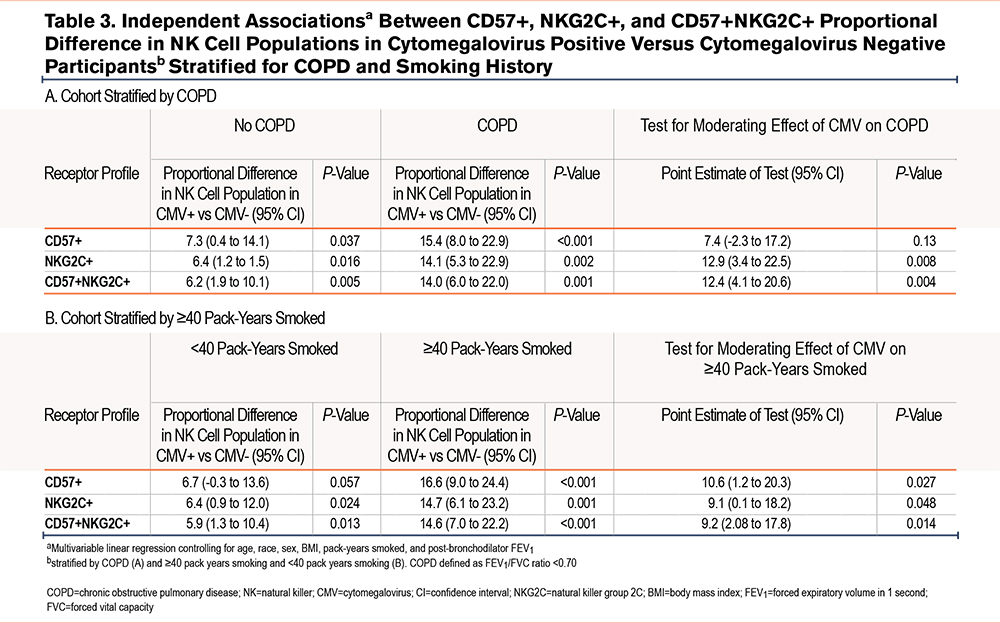
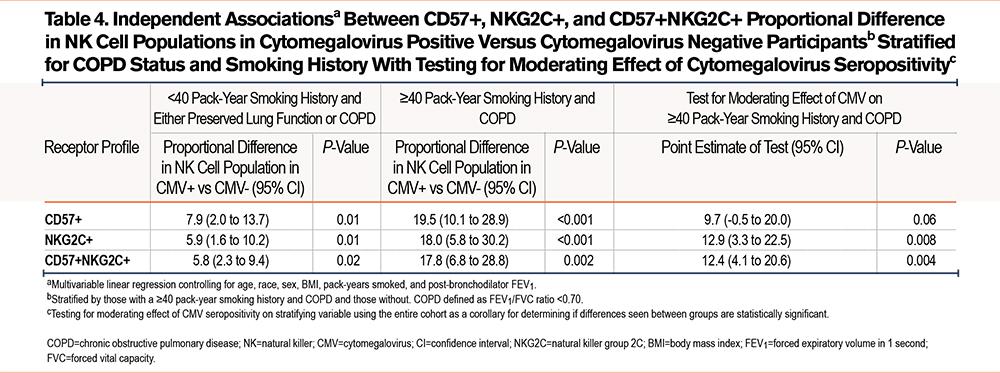
When stratified for COPD (Table 3), there was a numerical difference in the %-population of CD57+ NK cells in CMV seropositive participants in those without airflow limitation in (7.3 %-population [95% CI 0.4-14.1]; P=0.037) and those with COPD (15.4 %-population [8.0-22.9]; P<0.001). However, when checking the interaction term of COPD and CMV serostatus, CMV serostatus did not modify the relationship between COPD and %-population CD57+ NK cells. The relationship between CMV seropositivity and %-population CD57+ NK cells was only statistically significant in those who had a ≥40pack-year smoking history (16.6 %-population difference [95% CI 9.0-24.4]; P<0.001). CMV seropositivity did significantly modify the relationship between pack-years smoked groups and %-population CD57+ NK cells.
In those with COPD and a ≥40 pack-year smoking history versus those that did not meet these criteria and controlled for age, race, sex, and post-bronchodilator FEV1, heavy smokers with COPD who were CMV seropositive had a 19.5% difference in the proportion of CD57+ NK cells (95% CI 10.1 to 28.9; P<0.001) while those who did not meet these criteria had a mean difference in the CMV seropositive group of 7.9% (95% CI 2.0 to 13.7; P=0.01) (Table 4). In the entire cohort, CMV seropositivity did significantly modify the relationship between %-proportion CD57+ NK cells and those who both had COPD and reported ≥40 pack-years smoking.
NKG2C+ NK Cells
There were no relationships seen between the proportions of NKG2C+ NK cells and continuous covariables. Only CMV serostatus (10.6% difference in proportion) is associated with higher mean proportions of NKG2C cells among the total NK-cell population.
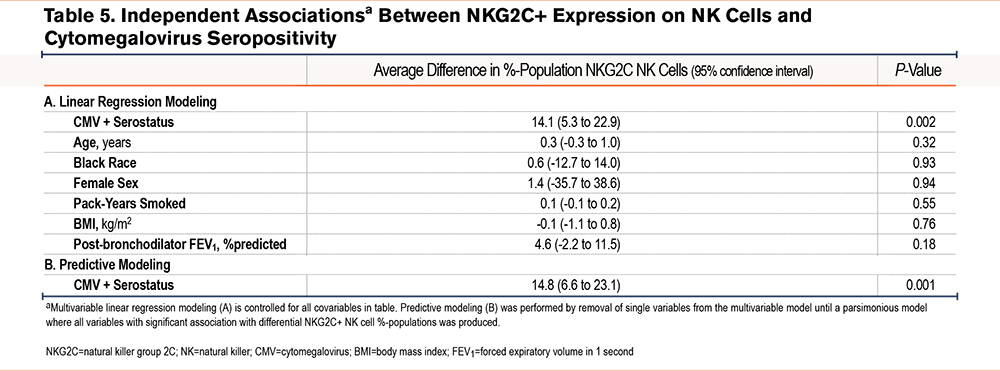
When controlled for clinically relevant covariables, CMV seropositive participants had a 14.1 higher %-population (95% CI 5.3 to 22.9); P=0.002) NKG2C+ cell population (Table 5). Similar to CD57+ models, we removed non-significant covariables to assess those that predict differential NKG2C+ populations. In this predictive modeling, only CMV seropositivity predicted a statistically significant difference in proportion of NKG2C+ cells among the NK cell population.
When stratifying for COPD, the independent relationship between CMV seropositivity and proportional NKG2C+ NK cell populations was statistically significant in both those without COPD (6.4 %-population difference [95% CI 1.2 to 11.5]; P=0.016) and those with COPD (14.1 %-population difference [95% CI 5.3 to 22.9]; P=0.002) (Table 3). CMV seropositivity did statistically modify the relationship between COPD and %-population of NKG2C+ NK cells. Likewise, CMV seropositivity was independently associated with a higher NKG2C+ NK cell population in those who smoked <40 pack years (6.4 %-population difference [95% CI 0.9 to 12.0]; P=0.024) and those with ≥40 pack-years smoking (14.7 %-population difference [95% CI 6.1 to 23.2]; P=0.001). CMV seropositivity significantly modified the statistical relationship between smoking ≥40 pack years and a higher proportion of NKG2C+ NK cells.
Differences in NKG2C+ NK cell populations associated with CMV serostatus between heavy smokers with COPD and those without were assessed (Table 4). Those with ≥40 pack-years smoking and COPD had an 18.0% higher mean proportion of NKG2C+ NK cells (95% CI 5.8 to 30.2; P<0.001) while, in those that did not meet these criteria, CMV seropositivity was associated with a 5.9% higher mean NKG2C+ NK cell proportion (95% CI 1.6 to 10.2; P=0.01) (Table 4). CMV seropositivity significantly modified the statistical relationship between heavy smoking and COPD and %-population NKG2C cells.
CD57+NKG2C+ NK Cells
In bivariable analysis, there were no continuous variables associated with double positive NK cells. However, COPD (4.8% difference in proportion of double positive NK cells among the population) and positive CMV affinity (10.5% difference) were associated with a higher percentage proportion of double positive cells among the NK cell population.
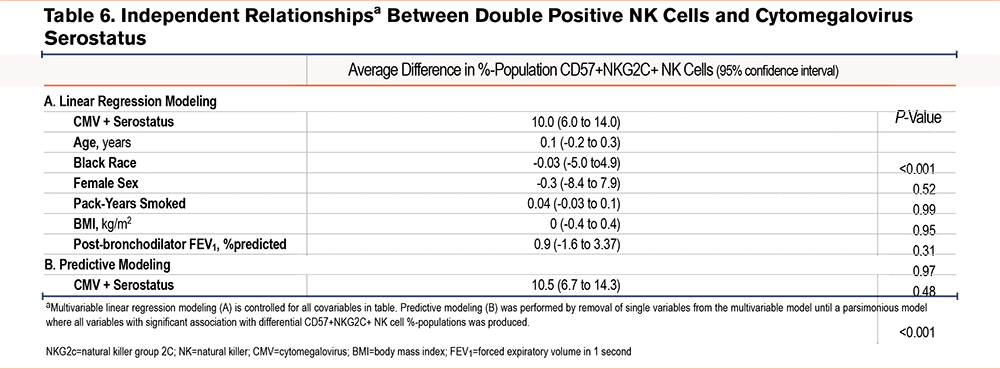
When controlled for clinically relevant covariables, CMV seropositivity was associated with a 10.0% higher proportion of double positive cells in the NK cell population (95% CI 6.0 to 14.0; P<0.001). We also performed predictive modeling for predictors of double positive cells and found, similar to NKG2C+ populations, only CMV affinity predicts differential populations (see bivariable analysis for associations) (Table 6).
When we stratified COPD and controlled for clinically relevant covariables (Table 3), CMV+ was associated with a proportionally higher %-population of CD57+NKG2C+ NK cells in both those with COPD (14.0 %-population difference [95% CI 6.0-22.0]; P=0.001) and without (6.2 %-population difference [95% CI 1.9–10.1]; P=0.005). CMV seropositivity significantly modified the relationship between COPD and %-population of CD57+NKG2C+ NK cells. When stratifying by ≥40 pack-years smoking and <40 pack years, CMV seropositivity was associated with a higher %proportion of CD57+NKG2C+ NK cells in both a heavy smoking history (14.6 %-population [7.0–22.2]; P<0.001) and <40 pack-years smoking (5.9 [95% CI 1.3-10.4]; P=0.004). In the entire cohort, CMV seropositivity significantly modified the relationship between %proportion CD57+NKG2C+ and pack-years smoked.
We assessed the association between double positive populations and CMV serostatus when stratifying by COPD and a ≥40 pack-year smoking history (Table 4). When controlled for clinically relevant covariables, those with COPD and heavy smoking and a positive CMV serostatus had a 17.8% higher proportion of double positive NK cells (95% CI 6.8 to 28.8; P=0.002). In those that did not meet these criteria, CMV+ participants have a 5.8% higher population proportion of double positive NK cells (98% CI 2.3 to 9.4; P=0.02). In the entire cohort, CMV seropositivity significantly modified the relationship between heavy smoking and COPD with %-population double positive cells.
Discussion
This analysis defines the relationships between CMV seropositivity and NK cell populations in a cohort of smokers with and at risk for COPD. In this cohort, we demonstrate that CMV seropositivity is associated with increased CD57+ and NKG2C+ populations supporting the known literature.19,23 Further, in this cohort we describe CMV serostatus as the primary predictor of higher proportions of NKG2C+ NK cells while smoking and higher BMI predicts higher proportions of CD57+ NK cells along with +CMV serostatus. The association with CMV seropositivity was greater in those with a heavy smoking history and COPD. When testing these numerical differences by assessing the moderating effect of CMV seropositivity and our strata mentioned above, CMV seropositivity significantly influences the relationship between all strata of interest for all NK cell populations except for the relationship between CD57+ cells and COPD and those with a heavy smoking history and COPD. The findings suggest that that CMV exposure/latent infection may be a specific factor influencing NK cell population in need of further investigation.
Our lab has previously described CD57+ and NKG2C+ NK cells making up a larger proportion of NK cell populations in smokers.12 In this same study, NK cells expressing these receptor profiles were found to make up approximately 80% of IFN-γ-producing NK cells. However, equipoise remains surrounding the effect of cigarette smoke on NK cell activity, as other studies describe NK cells extracted from smokers with less inflammatory potential than from non-smokers.24,25 These studies do not characterize receptor profiles. A future direction would be to characterize NK cell receptor shifts over time, if inflammatory potential of the NK cell population changes along with population phenotypes, and clinical manifestations associated with population dynamics. Defining the accrual of pro-inflammatory potential and associations with clinical outcomes is further supported by a cross-sectional, in vitro study that suggests that NK cell populations of smokers demonstrate a high-degree of natural cytotoxic potential directed at airway cells.13 While smoking has been shown to associate with NK cell population profiles,12 CMV remains another potential influencing factor.26 This study suggests that CMV seropositivity is associated with greater expression of CD57+ and NKG2C+ NK cells and that this association is stronger in those with COPD, those with a ≥40 pack-year smoking history, and those with both a heavy smoking history and COPD with 2 exceptions being CMV+ seropositivity does not modify the relationship between COPD and heavy smoking and COPD and CD57+ expression. This may be the result of CD57+ expression being uncoupled from emergence of other receptors that arise due to frequent stimulation, represent populations that have been frequently stimulated by multiple causes, and the biological importance of the CD57+/CD57- NK cell balance.27 This is juxtaposed with NKG2C+ cells which are thought to expand directly in response to CMV infection.15 This study is a cross sectional analysis and does not imply causation, however, our findings support further investigation of how smoking, COPD, and viral exposures shape the NK cell population, the inflammatory potential of these populations, and the influence of NK cell population dynamics on longitudinal COPD outcomes.
Our results suggest that, in cross-sectional analysis, CMV is associated with the emergence of CD57+ and NKG2C+ NK cell populations and suggests that CMV seropositivity is associated with emergence of a larger proportion of cells expressing these NK cell receptors in those with a heavy smoking history and COPD. While a summation of the evidence would suggest the expression of NK cell population phenotypes is multifactorial,12,15,26,27 this study supports a conceptual framework wherein exposure to a chronic CMV infection may differentially impact %-proportion of specific NK cell receptor profiles. While our findings are only correlative and do not imply mechanism, there have been findings that support further pursuit of the influence of CMV on NK cell population dynamics in COPD. Factors inherent to the inflammatory state of COPD may potentiate the effect of CMV on NK cells. COPD is well documented as a proinflammatory disease and smoking promotes inflammation.28 High levels of IL-12 and IL-18 activating cytokines may lead to expansion of NK cells29,30 along with differential expression of HLA-C and KIR in the chronically stressed pulmonary immune compartment in COPD.31 We purport a general hypothesis that the chronic stress of a latent CMV infection influences NK cell populations which in turn may lead to an overexuberant immune response in COPD. This study provides a platform for future investigation of how NK cell populations change over time, if this is associated with increased inflammatory potential, factors that lead to these changes, and if the differential population profiles and in vitro inflammatory activity is associated with differential clinical outcomes in COPD. Future studies may expand upon these findings with longitudinal study of environmental and infectious factors that lead to NK cell population change and increased inflammatory potential in smokers with COPD and associated clinical outcomes.
There is a paucity of information regarding the interaction of chronic CMV infection in chronic airways disease, the natural course of the infection (e.g., does an infection in childhood portend worse outcomes than an infection as an adolescent or adult), and any biological processes affected by chronic CMV infection. One study on adolescents with perinatal HIV in sub-Saharan Africa shows CMV increases the odds of having reduced lung function.32 This study is limited by the potential for protopathic bias (as are all studies in CMV and lung disease) and also by residual confounding as certain aspects of HIV and lung disease including more rapid deterioration in lung function is a response to environmental irritants and smoking33,34 as well as a higher risk of a multitude of lung infections35 that can lead to similar outcomes.
This study adds to the few foundational studies on how CMV influences the immune response in COPD and there is a need to better understand the impact of latent viruses in COPD. Studies supporting future directions have been published. In a cohort of Veterans who smoke, positive CMV serostatus was associated with greater odds of having prevalent airflow limitation when controlled for clinically relevant covariables.21 One study used Cox regression modeling in a retrospective study to show that CMV+ participants, specifically those less than 55 years old, had a higher risk of all-cause and COPD-related mortality than those without CMV.14 Along with epidemiological correlations, few studies provide foundational data on how CMV impacts the cellular immune system in COPD. CMV reactivation events have been posited to lead to system inflammation via the induction of CD28-null T-lymphocytes leading to a hyperinflammatory state that may hasten lung function decline in COPD.36 Further, along with smoking, chronic CMV infection may influence population dynamics and inflammatory potential of another innate lymphocyte, mucosal-associated invariant T cells,26,37,38 which also provide defense against bacteria commonly associated with COPD, most notably non-typeable Haemophilus.39 We report cross-sectional associations between CMV, smoking, COPD, and NK cell populations that support future investigations elucidating the interface of these factors over time and associated clinical manifestations. Uncovering meaningful clinical outcomes associated with NK cell activity is important to the field of COPD as current16,17 and emerging18 COPD therapies can potentially be used to target NK cell biology.
This study has several limitations. As this is a VA cohort, it is predominantly male. Further, as a cross-sectional study, we are only able to show correlations. Our study did not have a large enough enrollment to be adequately powered to describe CMV serostatus based on binding avidity, which would give a rough approximation of when a participant developed a CMV infection. Our approach to testing the influence of COPD in stratified cohorts involves the use of a statistical interaction term to determine the statistical influence of CMV serostatus between strata. This is not a direct statistical test for assessing difference in CMV associations between strata but does have a strength over standard effect size tests which are potentially nebulous and based on interpretation. Stratifying by smoking status within COPD strata would have been an informative analysis but our cohort is likely too small to produce valid outcomes from this approach. Sensitivity analyses incorporating general markers of inflammation would have supported our findings further. Also, characterizing inflammatory activity would give a deeper understanding of NK cell biology in this clinical cohort.
In conclusion, we describe an association between CMV seropositivity and NK cell populations in a cohort of smokers at-risk for and with COPD and suggest that the effect of CMV seropositivity on the association of CD57+ and NKG2C+ NK cell populations may be larger in those who smoke and have COPD, with a few exceptions for CD57+ expression. Our findings support future longitudinal studies elucidating viral and environmental factors of NK cell population make-up, other receptors or receptor clusters that emerge in response to smoking and CMV, the inflammatory potential of these populations, and the impact on clinical outcomes in COPD.
Acknowledgements
Author contributions: RMB, MTB, and RP designed the experiments and wrote the manuscript. EB, RMB, TH, and AO performed the experiments and data analysis. LL collected specimens, performed spirometry, and data collection. SNW aided in interpretation and writing of the manuscript. All authors reviewed and approved the manuscript for publication.
Declaration of Interest
The authors have declared that no conflict of interest exists.