Running Head: CELA1 in Alpha-1 Antitrypsin Deficient Emphysema
Funding Support: Funding for this work was provided by the Innovation Fund/Innovation Ventures of Cincinnati Children's Hospital, an Alpha-1 Foundation Research Award 498262, and the National Institutes of Health (NIH)/National Heart, Lung, and Blood Institute (NHLBI) grants (R01HL141229, and K08HL131261) provided to BMV. Additional funding was provided by NIH/NHLBI grant R01HL119538 and Veterans Administration grant I01BX002347 to MTB and by NIH/NHLBI grant U01HL148860 provided to JNA.
Date of Acceptance: July 18, 2023 | Publication Online Date: August 1, 2023
Abbreviations: AAT=alpha-1 antitrypsin; AGC=automated gain control; ANOVA=analysis of variance; CELA1=chymotrypsin-like elastase 1; CS=cigarette smoke; LC= liquid chromatography; LPS=lipopolysaccharide; LD-PPE=low-dose porcine pancreatic elastase; H&E=hematoxylin and eosin; MS=mass spectrometry; PBS=phosphate buffered saline; PPE=porcine pancreatic elastase; SPE=solid-phase extraction; WT=wild-type mice
Citation: Devine AJ, Smith NJ, Joshi R, et al. Chymotrypsin-like elastase-1 mediates progressive emphysema in alpha-1 antitrypsin deficiency. Chronic Obstr Pulm Dis. 2023; 10(4): 380-391. doi: http://doi.org/10.15326/jcopdf.2023.0416
Online Supplemental Material: Read Online Supplemental Material (5699KB)
Introduction
Alpha-1 antitrypsin (AAT) deficiency is a rare disease affecting approximately 1 in 2000 White individuals with approximately 10% of these individuals developing AATD lung disease.1,2 This lung disease is marked by progressive alveolar loss despite the withdrawal of triggering agents such as cigarette smoke.3 Although the PiZZ genotype is the most common mutation, there are over 100 described variants making true population estimates difficult4 and AAT deficiency is typically diagnosed by reduced levels of AAT in the blood. Typically developing in the fourth and fifth decades of life, AAT-deficient lung disease is marked by progressive emphysema and is the fourth leading indication for lung transplantation.1 AAT augmentation therapy does not prevent disease progression5 making the development of new therapeutic approaches critical.
Chymotrypsin-like elastase 1 (CELA1) is a serine protease synthesized and secreted by alveolar type 2 cells with a physiologic role in reducing postnatal lung elastance.6 At a molecular level, CELA1 binds and cleaves non-crosslinked, hydrophobic domains of tropoelastin, and its binding to lung elastin fibers is increased with strain—similar to other pancreatic elastases.7 CELA1 is neutralized by covalent binding with AAT, and Cela1-/- mice were completely protected from emphysema in an antisense oligonucleotide model of AAT-deficient emphysema.6 This model, however, did not include any injury apart from administration of the antisense oligonucleotide with levels of emphysema exceeding that seen in mice with genetic ablation of 5 Serpina1 paralogues and subjected to tracheal lipopolysacharide or cigarette smoke.8 Here, we use this murine genetic model of AAT deficiency to test the role of the CELA1 gene in AAT-deficient emphysema using multiple models to show that CELA1 has a role in progressive airspace enlargement in AAT-deficiency independent of inflammation.
Materials and Methods
Animal Care and Use
Animal use was approved by the Cincinnati Children's Hospital Institutional Animal Care and Use Committee (#2020-0054) with food and water provided ad-lib with 12-hour day/night cycles. For all experiments, approximately equivalent numbers of C57BL/6 male and female mice were used. Cela1-/-6 and AAT-deficient (Serpina1em3Chmu/J, JAX Stock# 035015) mice were used and genotyped per published protocols.8
Tracheal Porcine Pancreatic Elastase Model
In 8–10-week-old mice, 0.2 to 10 units of porcine pancreatic elastase (PPE) (Sigma #E1250), diluted to 20 units or 2 units per mL in phosphate-buffered saline [PBS]) was administered to mice after anesthetization with 2% isoflurane, suspension by the incisors on a rodent intubating board, tracheal cannulation with a 22-gauge angiocatheter, and administration of PPE during inspiration. Mice were recovered and sacrificed at specified time intervals after intraperitoneal administration of ketamine/xylazine/acepromazine 70/5/2mg/kg followed by exsanguination and vital organ harvest.
Cigarette Smoke Exposure Model
Beginning at 10–12 weeks of age, mice were exposed to 4 hours of whole-body cigarette smoke 5 days per week for 10 months using a Teague TE-10z smoking machine using smoke generated from 3R4F Kentucky Reference Cigarettes (University of Kentucky) at a concentration of 150 mg/m3 total suspended particulates. This work was performed under approval from the Institutional Animal Care and Use Committee at the University of Cincinnati Medical Center Protocol# AM06-20-10-28-01.
Aging Model
Male and female mice were allowed to age to 72–75 weeks and sacrificed as above.
Lung Tissue Processing and Analysis
After exsanguination, a neck incision was made and the tracheal cannula secured with a 3-0 silk suture around the trachea, the left bronchus was ligated with a surgical clip, and the left lung snap frozen, and the right lung was inflated with 2% paraformaldehyde in PBS at 20 cm water pressure. The trachea was ligated with the removal of the cannula, fixed overnight in 2% paraformaldehyde dehydrated to 100% methanol, paraffinized, and right lung lobes randomly oriented, embedded, sectioned at 5µm, and mounted. Sections were hematoxylin and eosin (H&E) stained and up to five 10X images were obtained per lung lobe. Mean linear intercepts of blinded sections were determined using the methods of Dunnill.9 Airspace diameters of tile-scanned 4X sections were determined using a previously described MatLab (MathWorks, Natick, Massachusetts) code.10
Biochemical Assays
The upper half of snap frozen left lung lobes was homogenized in PBS and myeloperoxidase activity was quantified using published protocols.11
Proteomic and Bioinformatic Analysis
Frozen left lung lobes were cut in half, proteins were extracted using the MPLEx methods as previously described.12 For each sample, extracted proteins were denatured, alkylated, digested with trypsin, and desalted on a C18 solid-phase extraction (SPE) cartridge (Discovery C18, 1ml, 50mg, Sulpelco). The peptide concentration was measured by bicinchoninic assay (ThermoScientific). Samples were analyzed using a label-free liquid chromatography (LC)-mass spectrometry (MS)/MS strategy. Briefly, 5μl of samples at the concentration of 0.1μg/μL were loaded on an SPE column and separated on a reverse phase C18 analytical column using a 180-min gradient.
MS data were recorded 60 min after injection. The effluents from the LC column were ionized by electrospray ionization by applying 2200 V to the electrospray tip. Electrosprayed ions were introduced into the mass spectrometer (Orbitrap Q Exactive plus) via a heated capillary. The resulting ions were mass analyzed by the Orbitrap at a resolution of 70,000 covering the mass range from 300 to 1800 Da with a maximum injection time of 20 ms and automated gain control (AGC) setting of 3e6 ions. Most abundant ions were subjected to MS2 analysis using a top 12 strategy (i.e., top 12 ions are fragmented during between 2 precursor scans). For MS2, ions with charge states different than 1 were isolated by quadrupole mass filter in monoisotopic peak selection mode using an isolation window of 1.5 Da, maximum injection time of 50 ms with AGC setting at 1e5 ions and fragmented by high-energy collision dissociation with nitrogen at 30% normalized collision energy. Fragment ions were mass analyzed by the Orbitrap at a resolution of 17,500 covering the mass range from 200 to 2000 Da, and spectra were recorded in the centroid mode. Ions once selected for MS2 were dynamically excluded for the next 30 seconds.
Raw mass spectrometry data was searched using MaxQuant (v1.6.0.16) and LFQ data were analyzed using RomicsProcessor13 (code DOI:10.5281/zenodo.3386527). Data were filtered for unique proteins being detected in at least 50% of samples and then log2 transformed and median normalized. Missing values were imputed using a downshifted randomly distributed data14 analysis of variance (ANOVA) and Welch’s t-tests were performed (2-tailed, unequal variance). Protein-protein interaction networks were generated using the STRING database.15 Volcano plots were created using EnhancedVolcano.16 Proteomic datasets are available at MassIVE (Accession: MSV000091330).
Statistical Analysis
Comparisons were made using Welch’s t-test or ANOVA with Holm-Sidak post hoc comparison as appropriate using rstatix17 using R-4.1.0.18 Plots were created using ggpubr19 with central lines indicating mean and whiskers one standard deviation. P-values of <0.05 were considered statistically significant.
Results
CELA1 in Tracheal Lipopolysaccharide Model Alpha-1 Antitrypsin-Deficient Emphysema
Borel et al8 previously reported that AAT-deficient mice developed more emphysema at 14 days than wild-type mice after tracheal doses of 1 and 0.5 units of lipopolysaccharide at days 1 and 10. We intended to test whether Cela1-/- &AAT-deficient mice were protected from emphysema in this model. However, we observed a similar degree of emphysema in wild-type and AAT-deficient mice (Figure 1A), with male and female mice similarly affected. We then performed a dose titration experiment with 1 and0.5, 2 and1, 5 and2.5, and 10 and5 units of LPS with evaluation at 21 days instead of 14 and evaluated airspace sizes using a previously described method that generates descriptive statistics of airspace sizes in tile-scanned images of all lobes.6,10 There was no clear dose effect and wild-type mice experienced as much or more emphysema as AAT-deficient mice (Supplemental Figure 1 in the online supplement). Since we could not reproduce a previously reported LPS-based model of AAT-deficient emphysema, we developed a different model of AAT-deficient emphysema.
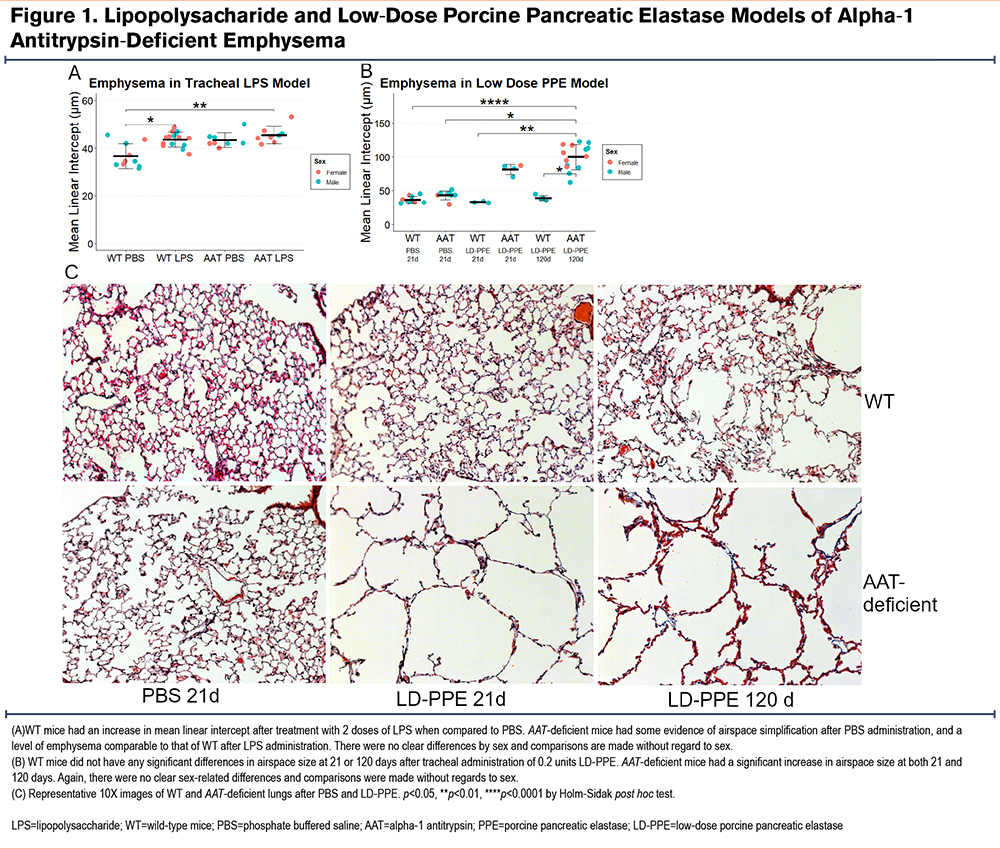
Low-Dose Porcine Pancreatic Elastase Model of Alpha-1 Antitrypsin-Deficient Emphysema
Tracheal PPE is a well-established model that causes significant injury and, in some studies, progressive emphysema.20,21 In most studies, 1–2 units of PPE are administered tracheally, and emphysema is assessed at or before 21 days. We administered 2 units of PPE to AAT-deficient mice, and 6 of 8 mice died with extensive lung hemorrhage on necropsy. We performed a dose titration experiment and discovered that 0.2 units of tracheal PPE caused substantial emphysema in AAT-deficient mice at 21 days while wild-type mice had virtually no emphysema at this dose. Furthermore, emphysema progressed over time (Figure 1B and C and Supplemental Figure 2 in the online supplement). We, therefore, administered 0.2 units of tracheal PPE to AAT-deficient and Cela1-/- &AAT-deficient mice to test for a role of the CELA1 gene in emphysema in the context of AAT-deficiency.
Lung Proteomic Changes in Low-Dose Porcine Pancreatic Elastase Model
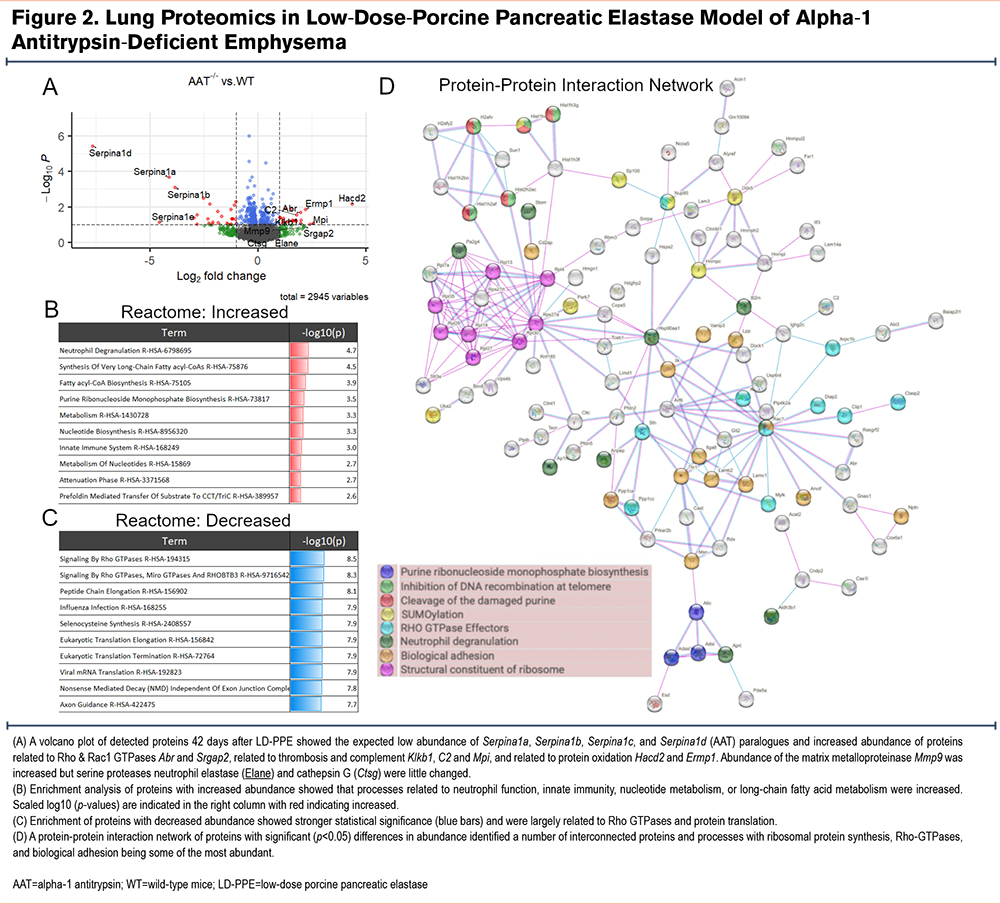
We performed unbiased proteomics of wild-type, AAT-deficient, Cela1-/- and Cela1-/- &AAT-deficient lungs (5 per genotype) 42 days after low-dose PPE to understand the differential response to injury. In comparing wild-type and AAT-deficient lungs, only one protein had an adjusted p-value<0.1 and at least a 2-fold change. This protein was the AAT isoform Serpina1d which is one of the 5 AAT paralogues deleted in this mouse.8 Seventeen genes had protein levels at least 2-fold lower in AAT-/- compared to wild-type lungs, and 71 genes were at least 2-fold higher (Supplemental Table 1 in the online supplement). As expected, many of the most downregulated genes in this analysis were AAT paralogues (e.g., Serpina1a, Serpina1b, and Serpina1d). Genes of many of the upregulated proteins were related to Rho and Rac1 GTPases (Abr and Srgap2), thrombosis and complement (Klkb1, C2 and Mpi), and protein oxidation (Hacd2 and Ermp1) (Figure 2A). Gene ontogeny enrichment for upregulated proteins identified neutrophil degranulation, functions related to innate immunity, and nucleotide biosynthesis pathways as the most activated (Figure 2B). The statistical significance of downregulated pathways was greater and largely involved processes related to mRNA translation (Figure 2C). The protein-protein interaction network of both increased and decreased proteins showed many edges related to ribosomes and translation, and that many of the proteins related to neutrophil degranulation were either not adjacent in the network or not within the protein-protein interaction network at all (Figure 2D and Supplemental Figure 3 in the online supplement). These data are consistent with the expected pulmonary biology of AAT-deficiency with regards to AAT synthesis and innate immunity but also some unexpected changes with regards to protein synthesis.
CELA1 in Low-Dose Porcine Pancreatic Elastase Model of Alpha-1 Antitrypsin-Deficient Emphysema
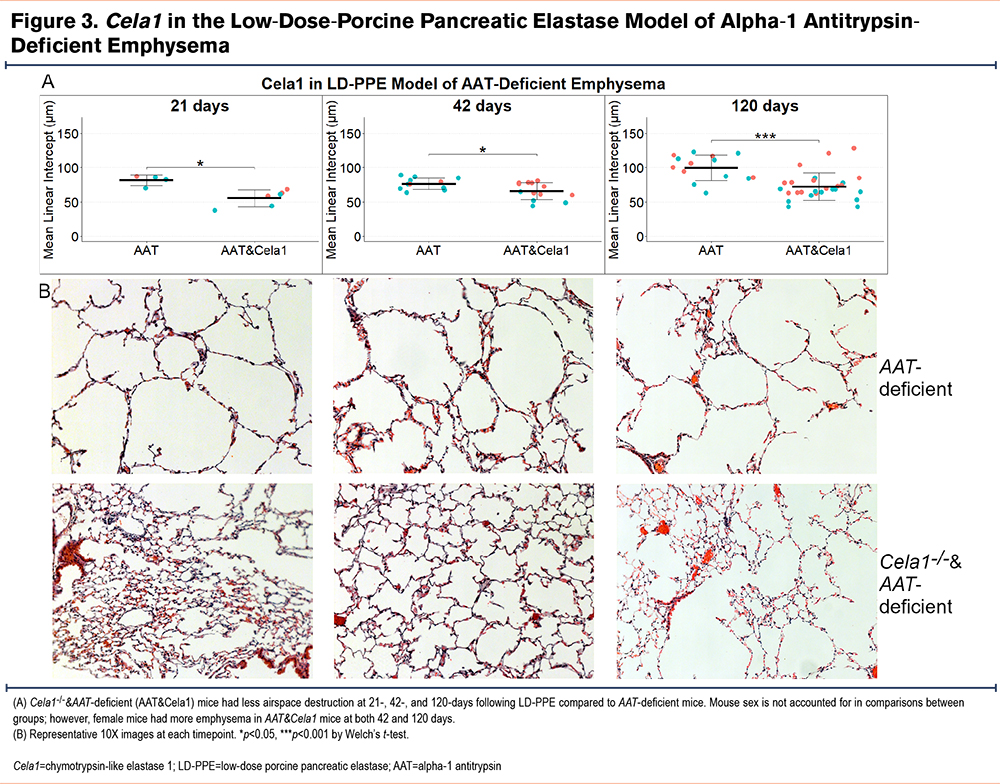
We tested for a role for CELA1 in progressive emphysema by quantifying mean linear intercepts at 21, 42, and 120 days after low-dose PPE in AAT-deficient and Cela1-/- &AAT-deficient mice. Compared to AAT-deficient mice, Cela1-/- &AAT-deficient mice had less emphysema at 21, 42, and 120 days after low dose PPE administration (Figure 3A and B). Overtime, AAT-deficient but not Cela1-/-&AAT-deficient mice experienced progressive emphysema; between days 42 and 120, AAT-deficient mice had a 31% increase in mean linear intercept (p=0.002) while the increases in this groups between 21 and 42 days or and in the Cela1-/-&AAT-deficient mice over time were smaller and not significant. We observed that at 42 and 120 days, female Cela1-/-&AAT-deficient but not female AAT-deficient mice had more emphysema than male mice (p=0.02 and p=0.01 respectively). However, when analyzing by sex, the 120-day mean linear intercept differences remained significant for males (p=0.003) and females (p=0.03). These data indicate that CELA1 plays a role in AAT-deficient emphysema and perhaps this role is more pronounced in males than females.
Impact of CELA1 on Lung Proteome in Low-Dose Porcine Pancreatic Elastase Model
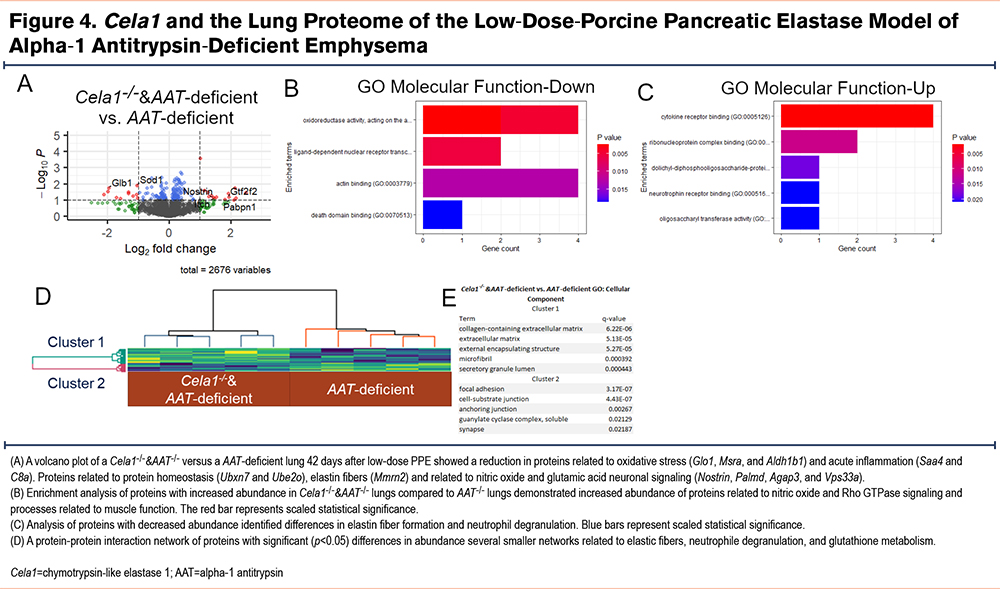
We then compared the lung proteomes of Cela1-/-&AAT-deficient and AAT-deficient mice 42 days after tracheal administration of low-dose PPE. There were only 2 proteins with adjusted p-values of less than 0.1 and at least a 2-fold change. The genes of these proteins were UBX domain-containing protein 7 and Ig alpha chain C region. There were 19 proteins that were at least 2-fold increased and 28 proteins that were at least 2-fold decreased in Cela1-/-&AAT-deficient compared to AAT-/- mice (Supplemental Table 2 in the online supplement). Among the proteins with increased abundance, some of the most significant were from genes related to oxidative stress (Glo1, Msra, and Aldh1b1) and acute inflammation (Saa4 and C8a). Proteins that were most increased were from genes related to ubiquitination (Ubxn7 and Ube2o), elastin fibers (Mmrn2), and neurotransmitters (Nostrin, Palmd, Agap3, and Vps33a) (Figure 4A). Gene ontogeny enrichment for proteins that were increased showed abundance of Rho GTPase proteins, nitric oxide signaling, and muscle-related proteins (Figure 4B). Downregulated processes included elastic fiber formation and neutrophil degranulation (Figure 4C). Protein-protein interaction analysis of increased and decreased proteins identified a number of small but connected networks related to elastin fibers, neutrophil degranulation, and glutathione metabolism (Figure 4D). These data demonstrate that Cela1 mediates protein-level differences in elastin microfibril associated proteins in this AAT-deficient model of emphysema and that there are also potential differences in neutrophil activity and oxidative state.
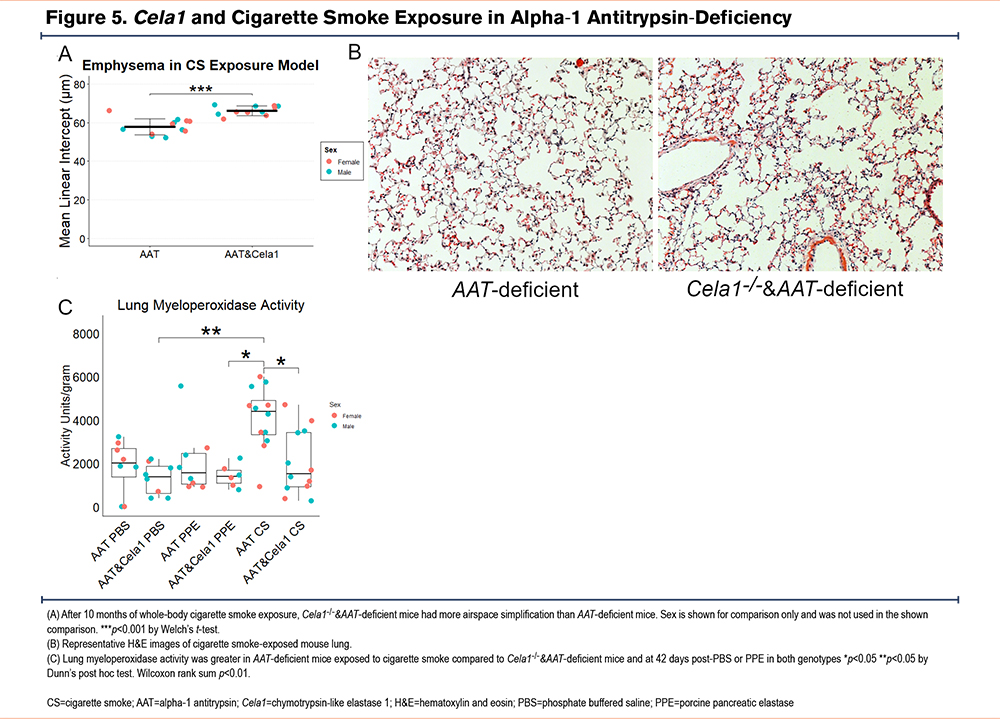
CELA1 in Cigarette Smoke-Induced Emphysema
As cigarette smoke exposure is highly associated with the development of emphysema in AAT-deficient individuals, we performed whole body cigarette smoke exposure to AAT-deficient and Cela1-/-&AAT-deficient mice 5 days a week for 10 months. Contrary to our expectations, Cela1-/-&AAT-deficient mice were not protected from cigarette smoke-induced emphysema and Cela1-/-&AAT-deficient mice had more evidence of airspace destruction than AAT-deficient mice (Figure 5A and B). Given the proteomic findings of altered neutrophil-associated and chemotaxis proteins in Cela1-/-&AAT-deficient mice, we hypothesized neutrophil activity might be increased. However, myeloperoxidase activity was reduced by more than half in Cela1-/-&AAT-deficient mice compared to AAT-deficient mice in response to cigarette smoke and comparable to activity levels seen 42 days after PPE and PBS. (Figure 5C). While the mechanism is unclear, these data indicate the absence of Cela1 enhances airspace destruction in AAT-deficient mice response to sustained cigarette smoke exposure.
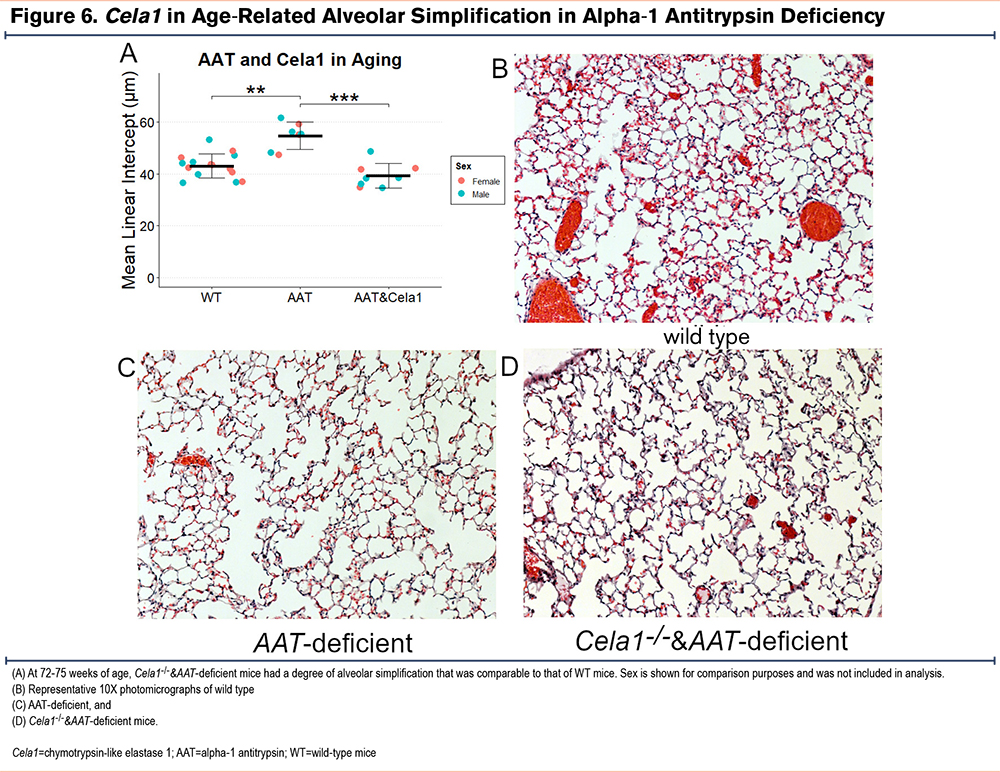
CELA1 in Age-Related Alveolar Simplification
In humans and mice, aging is associated with slowly progressive airspace simplification, and AAT-deficient mice were reported to have more rapid simplification than wild type.8 We validated these findings and discovered that Cela1-/-&AAT-deficient mice experienced a level of age-related airspace simplification similar to that of wild-type mice (Figure 6).
Discussion
This is the first study to show in a novel mouse model of injury-induced emphysema in AAT-deficiency that Cela1 is required for progressive airspace enlargement, and that this gene is important in age-related alveolar simplification. Interestingly, Cela1 did not play a role in cigarette smoke-induced emphysema suggesting that the mechanism by which chronic inflammation and injury causes emphysema is independent of post-injury remodeling. When considered in the context of our previous work showing that Cela1 binds to lung elastin in the context of strain,6 these findings support the concept that altered local lung mechanics in emphysema promote CELA1-mediated remodeling leading to progressive emphysema and that the lack of neutralization of CELA1 protein by AAT exacerbates this process.
The 4 models used in our studies help elucidate the role of CELA1 in AAT-deficient emphysema. In our first experiment, we were unable to recapitulate the emphysema seen with LPS administration in AAT-deficient mice.8 Potential reasons for this could be differences in microbiome, reduced LPS potency, or differences in administration technique. The finding that increasing doses of LPS caused more airspace injury in wild type than in AAT-deficient mice supports the conclusion that some intrinsic difference between facilities (such as microbiome) is responsible. For our second experiment, we modified the tracheal PPE model for use in AAT-deficient mice. The observed pulmonary hemorrhage and lethality with standard dose of PPE in AAT-deficient mice is probably because after tracheal PPE administration, there is leakage of serum into the airspace allowing AAT to neutralize the proteases in PPE. In AAT-deficient mice, the proteases in PPE remain active for a longer period leading to more extensive damage and death. Low-dose PPE administration to AAT-deficient mice produced robust emphysema that was progressive—an important clinical feature of AAT-deficient emphysema. The cigarette smoke exposure model is more clinically relevant than the PPE model, but it is dissimilar to human AAT-related lung disease because while smoking cessation is a mainstay of treatment for human AAT-related lung disease, in the mouse model, exposure is continuous. Given the relatively small increase in mean linear intercept observed, cessation of cigarette smoke exposure experiments would need a large number of mice and are probably inappropriate for Cela1 studies since some consequence of its absence seems to worsen cigarette smoke-induced emphysema. Notably, Steams et al recently showed that AAT knockdown using antisense oligonucleotides increased Cela1 expression but did not worsen lung injury in a cigarette smoke and influenza model.22 Lastly, we validated previously reported findings regarding age-related alveolar simplification in AAT-deficient mice8 and showed that the gene CELA1 plays an important role in this process. In considering these findings in the context of the physiologic role of CELA1—namely the reduction of postnatal lung elastance6—data from these 4 models demonstrate that in the context of AAT-deficiency, CELA1 does not appear to be involved in the lung’s response to injury but is important for continued airspace destruction once emphysema is established.
This study adds to our understanding on the role of sex on AAT-deficient emphysema. We previously reported that in the antisense oligonucleotide model, AAT-deficient mice had more significant emphysema,23 but we did not see these same changes in the genetic model. In the current study, while Cela1 ablation reduced emphysema in both male and female AAT-deficient mice in the LD-PPE model, the protection was greater in male mice. Previous studies did not show any effect of sex on lung Cela1 expression.6 These findings suggest that CELA1 could play a role in the increased sensitivity of AAT-deficient female patients compared to male ones.24
The progressive nature of emphysema in mouse models is controversial. In mice, rats, and rabbits, studies show that emphysema improves,25 is stable,26-28 or progresses,20,21,29 over time. While comparison between studies of different species and doses of PPE are difficult, it does appear that studies with stable or improved mean linear intercept have lesser degrees of initial injury than those that show progression. The role of localized lung tissue mechanics in emphysema progression was convincingly demonstrated by Toumpanakis et al.30 Mice treated with PPE were followed to 21 days with subsequent reduction of tracheal cross-sectional area by 50% for 24 or 72 hours. Emphysema was substantially greater with 72 hours of airflow restriction.30 Perhaps there is threshold of alveolar injury beyond which localized tissue mechanics become too deranged to make re-alveolarization possible; a hypothesis that warrants future study as understanding key processes of repair-regeneration in emphysema could lead to novel therapies to restore lung function in emphysema. There is likely an age-related component to repair potential as well since older mice have less compensatory lung growth than younger ones.31 While in human emphysema, alveolar loss is considered permanent, the adult human lung has the potential to re-initiate alveolarization post-pneumonectomy32 lending hope to the idea that if emphysema progression could be halted, other therapies could be employed to regenerate damaged lung tissue.
Our study has several limitations. First, we did not determine if Cela1-/-&AAT-deficient mice had preservation of lung function or diffusing capacity, and we did not measure lung volume in a way that would have permitted calculation of alveolar surface area. Our aged mice were harvested at 15 months and not 18 or 24 months as in some studies. We decided on this time point after an unexpected mortality in an aged AAT-deficient mouse at 15 months and concern for losing additional mice. To the best of our knowledge, there are no reports of early mortality in AAT-deficient mice. We previously identified the CELA1 gene as being expressed in alveolar type 2 cells, but we did not provide any cell-specific information here. Lastly, while we showed a specific role for CELA1 in this process, we did not interrogate the role of other proteases or test whether AAT-replacement was sufficient to protect mice from emphysema in the various models tested. As AAT has anti-inflammatory properties,33 it may be that CELA1 inhibition and AAT replacement therapy could be complementary.
Conclusions
CELA1 mediates progressive airspace destruction in AAT-deficient emphysema and its inhibition may represent a novel therapy in AAT-related lung disease.
Acknowledgements
Author contributions: All authors substantially participated in the creation of the submitted work. AJD, MTB, GCC, JNA, and BMV were responsible for the conception and/or design of the study. AJD, NJS, RJ, QF, GCC, and BMV were responsible for theacquisition of the data and data analysis. AJD, NJS, RJ, QF, MTB, JNA, and BMV were responsible for interpretation of the data. AJD, NJS, RJ, MTB, GCC, JNA, and BMV were responsible for writing the article and gave substantial involvement in its revision prior to submission. All authors significantly contributed to the intellectual content of the article and gave final approval of the version to be published.
A portion of this research was performed in the Environmental Molecular Sciences Laboratory, a Department of Energy Office of Science User Facility sponsored by the Biological and Environmental Research program under Contract No. DE-AC05-76RL01830. We thank Matthew Monroe for assisting in submitting the raw mass spectrometry data to MassIVE.
Declaration of Interests
BMV and Cincinnati Children’s Hospital hold patent PCT/US2020/061774 titled “CELA-1 Inhibition for Treatment of Lung Disease.” None of the other auhors have any conflicts to declare.