Running Head: Phase 1 Study of INBRX-101 in Adults With AATD
Funding Support: Inhibrx, Inc., funded this study and contributed to its design. Data were analyzed and interpreted by Inhibrx, Inc., in collaboration with all the authors. Medical writing assistance was also funded by Inhibrx, Inc.
Date of Acceptance: April 15, 2024 | Publication Online Date: N/A
Abbreviations: AAT=alpha-1 antitrypsin; AATD=alpha-1 antitrypsin deficiency; ADA=antidrug antibody; AE=adverse event; AUC0-7 days,ss=steady-state area under the plasma drug concentration-time curve, days 0 to 7; BALF=bronchoalveolar lavage fluid; Cavg=average concentration; Cavg, ss=average concentration at steady state; Cmax=maximum concentration; Cmax, ss=maximum concentration at steady state; Ctrough, ss=trough concentration at steady state; CT=computed tomography; DLCO=diffusing capacity of lung for carbon monoxide; fAAT=functional alpha-1 antitrypsin; FEV1=forced expiratory volume in 1 second; NE=neutrophil elastase; PD=pharmacodynamics; pdAAT=human plasma–derived alpha-1 antitrypsin; PELF=pulmonary epithelial lining fluid; PK=pharmacokinetics; Q1W=once weekly; Q3W=every 3 weeks; Q4W=every 4 weeks; SD=standard deviation
Citation: Brantly ML, Kuhn BT, Farah HW, et al. Recombinant alpha-1 antitrypsin–fc fusion protein INBRX-101 in adults with alpha-1 antitrypsin deficiency: a phase 1 study. Chronic Obstr Pulm Dis. 2024; 11(3): 282-292. doi: http://doi.org/10.15326/jcopdf.2023.0469
Online Supplemental Material: Read Online Supplemental Material (670KB)
Introduction
Alpha-1 antitrypsin deficiency (AATD) is a rare, chronic genetic disorder characterized by low serum levels of the serine protease inhibitor alpha-1 antitrypsin (AAT). In AATD, neutrophil elastase (NE) activity in the lungs is insufficiently controlled by AAT, which can result in progressive lung damage and pulmonary complications. Patients with severe AATD (e.g., SERPINA1 genotypes PiZZ, PiZ/Null, and PiNull/Null) have serum AAT concentrations <15%–20% of those in individuals with the wild-type genotype (PiMM); individuals with moderate deficiency (e.g., PiMZ and PiSZ) may have levels ≥49% of those in individuals with the PiMM genotype.1-5 Generally, lower serum AAT levels are correlated with a greater risk for pulmonary deterioration, poorer quality of life, and an increased mortality rate.5-10
The standard of care and only specific treatment available for AATD is augmentation therapy using human plasma−derived AAT (pdAAT) at 60mg/kg.2,10,11 With a mean half-life of 5−7 days,12-16 once-weekly infusion is required, creating a considerable burden for patients.8,17 Informed by studies from the 1980s, augmentation therapy with pdAAT aims to increase serum AAT levels above a theoretically “protective” threshold of 11µM.12,13,18-21 The relevance and validity of this threshold, which is approximately 50% below the lower limit of serum AAT levels in individuals with the PiMM genotype (range, 20−53µM),22 have been increasingly questioned.23-28 However, studies of higher dosing and the impact of elevating serum AAT levels further, including in patients with more moderate deficiency of serum AAT (e.g., PiMZ genotype), have historically been limited by the high cost of pdAAT and concerns about diverting limited supply from patients with severe deficiency.2,26,27,29
Evidence suggests that achieving higher AAT levels closer to those observed in PiMM individuals may lead to improved clinical outcomes. In the randomized, placebo-controlled trial RAPID, a post hoc analysis found an association between higher median trough levels of total serum AAT (90th percentile, >20µM) and a slower annual rate of lung density decline.30 Notably, this relationship between response and trough levels did not plateau, suggesting that higher serum AAT levels could further improve response. In a small study,31 a weekly double dose of pdAAT (120mg/kg), for which mean total serum AAT trough levels were within the range observed in PiMM individuals (20–53µM),22,32 led to greater reductions in bronchoalveolar lavage fluid (BALF) and plasma protease activity, BALF elastin degradation, and markers of airway inflammation compared with the standard dosing (60mg/kg weekly).
As total serum AAT levels with pdAAT 60mg/kg once weekly typically fall below 20µM (the lower limit of the range observed in PiMM individuals22) within 3−5 days of dosing,19,21,32 therapies are needed that lead to higher AAT levels with less frequent dosing. INBRX-101 is a recombinant human AAT-Fc fusion protein designed to overcome the limitations of pdAAT.33,34 INBRX-101 is composed of 2 recombinant human AAT molecules covalently linked to a human IgG4 Fc region and was optimized to have a longer half-life and higher therapeutic exposure (i.e., achieve higher functional AAT [fAAT] levels as measured by NE inhibition) than pdAAT (Figure 1A). The hinge and Fc regions have been modified for hinge stabilization, effector function silencing, and extended plasma half-life. Amino acid changes in the AAT reactive site loop mitigate loss of activity due to oxidation without impacting specificity for NE. In preclinical studies, INBRX-101 demonstrated NE inhibitory capacity comparable to that of pdAAT in vitro and in vivo.33 Importantly, INBRX-101 could penetrate the lungs in a murine model of pulmonary inflammation. Building upon these findings, a phase 1 study of INBRX-101 in patients with AATD was conducted. We present findings from the final analysis of this trial.
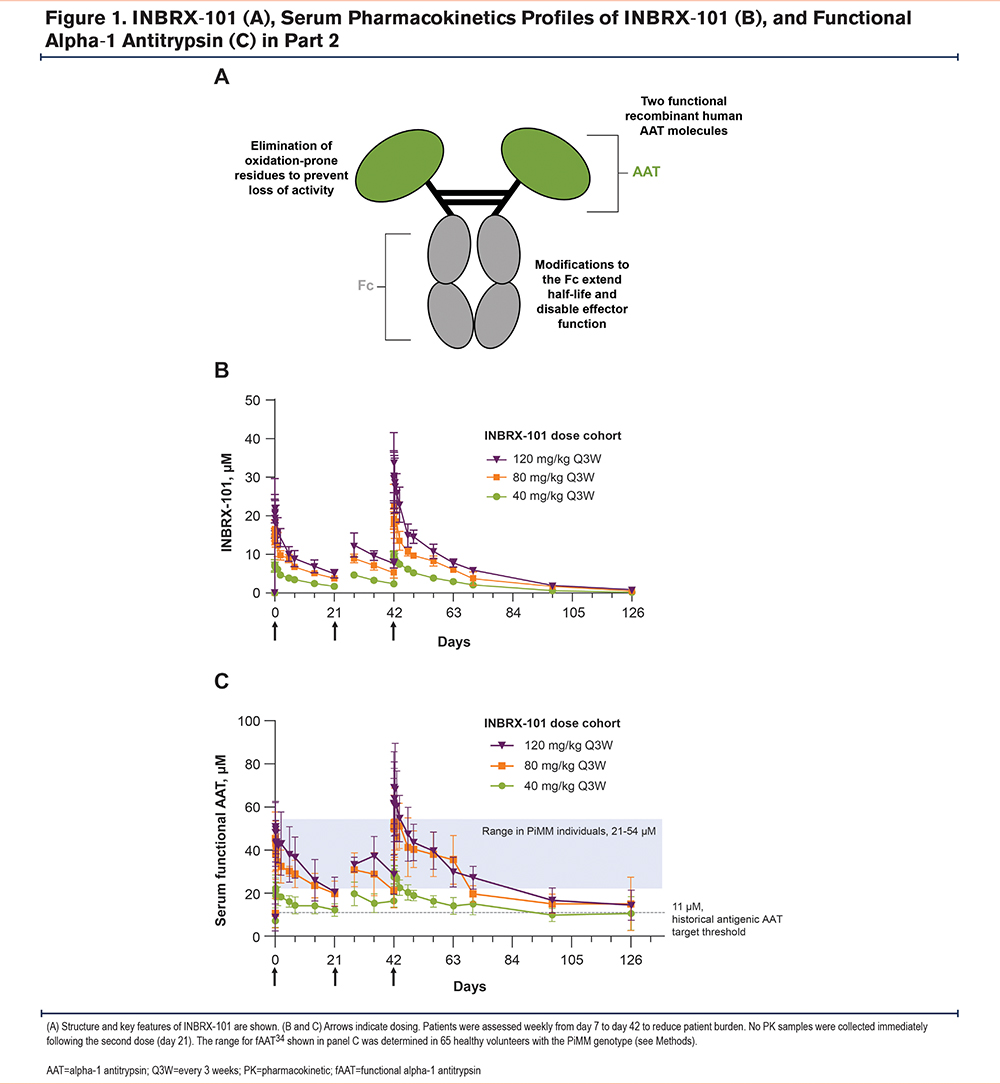
Methods
Study Design and Patients
This open-label, multicenter, 2-part, phase 1, dose-escalation study evaluated INBRX-101 in adults with AATD (Supplementary Figure 1 in the online supplement) (ClinicalTrials.gov identifier, NCT03815396). In part 1 (single ascending dose escalation), patients received 1 dose of 10, 40, 80, or 120mg/kg INBRX-101. In part 2 (multiple ascending dose escalation), patients received 3 doses of 40, 80, or 120mg/kg every 3 weeks (Q3W). Patients from part 1 could continue to part 2; participation in part 1 was not required for part 2. INBRX-101 was administered by intravenous infusion over 30 or 45 minutes.
The primary endpoint was safety and tolerability of INBRX-101 in patients who received ≥1 dose of INBRX-101 (safety population). Adverse events (AEs) were graded using the National Cancer Institute Common Terminology Criteria for Adverse Events, version 5.0, and coded using the Medical Dictionary for Regulatory Activities, version 20.1. Secondary endpoints included: (1) pharmacodynamics (PD) of fAAT in serum, (2) pharmacokinetics (PK) of INBRX-101 in serum and pulmonary epithelial lining fluid (PELF), (3) immunogenicity of INBRX-101, and (4) PD of fAAT in PELF. The PK population comprised all dosed patients with sufficient serum concentrations for PK evaluations; the PD population included dosed patients with ≥1 measurable PD value.
Eligible patients were aged 18–80 years (18–75 years in the 80- and 120-mg/kg cohorts in part 2) with a diagnosis of AATD (any allelic combination except those resulting in a null/null genotype) and previously documented antigenic serum AAT level of <11μM prior to starting pdAAT augmentation therapy. A washout period of ≥4 weeks before study dosing was required for patients receiving augmentation therapy; dosing could not be resumed for the duration of the study, including follow-up. Smoking was not allowed for the study duration, and patients had to be nonsmokers for ≥6 months. All patients provided written informed consent before participation. This study was conducted in compliance with the protocol, Declaration of Helsinki, the International Council for Harmonisation Guidelines for Good Clinical Practice, and applicable national and local laws and regulatory requirements. The protocol was approved by the independent ethics committee or institutional review board at each site (Supplementary Table 1 in the online supplement).
Procedures
Concentrations of INBRX-101 in serum samples were determined using a validated antigenic plate-based assay. A liquid chromatography multiple reaction monitoring mass spectrometry−based method was used to quantify INBRX-101 in human PELF. PK parameters were estimated via noncompartmental analysis using Phoenix WinNonlin version 8.3 (Certara, Radnor, Pennsylvania).
fAAT levels in serum and PELF were determined using validated anti-NE capacity methods. AAT calibrators (Prolastin-C), quality controls, and samples were incubated with a fixed amount of human NE; a fluorogenic NE substrate (N-methoxysuccinyl-Ala-Ala-Pro-Val-7-amino-4-methylcoumarin) was added and the fluorescent signal measured over 10 minutes. To estimate a preliminary range of fAAT in individuals with wild-type SERPINA1 genotype PiMM, serum samples from 65 healthy volunteers (verified as PiMM by the A1ALC test at Mayo Clinic Laboratories) were assayed using the same validated assay.
BALF was collected from patients in the 80- and 120-mg/kg cohorts in part 2 at screening and day 56 (i.e., 2 weeks after the third dose of INBRX-101); 3 lobes were sampled at each time point. Bronchoscopies were performed at 5 of the study sites by qualified medical staff per the Inhibrx-specified Bronchoscopy Procedure Manual; BALF was assessed by 3 specialized laboratories.
Immunogenicity to INBRX-101 and AAT were separately determined using traditional 3-tiered bridging electrochemiluminescence assays with sample pretreatment steps to improve tolerance to native AAT and INBRX-101. Characterization of antidrug antibody (ADA) domain specificity to the Fc/hinge portion of INBRX-101 was performed in a modified confirmatory tier using a domain-specific competitive reagent. Neutralizing antibodies to INBRX-101 and AAT were characterized using validated enzymatic activity assays.
Monolix 2021R1 and Simulx software (Lixoft, Antony, France) were used for population PK modeling and simulations. A 2-compartment model with linear elimination was used to describe INBRX-101 PK. Prediction-corrected Visual Predictive Checks and summaries of simulation results were created using RStudio (2022.02.3) and R (version 4.1.0). To simulate fAAT levels for phase 2 dose selection, a distribution of baseline fAAT values was generated based on the pooled mean (6.24µM) and standard deviation values (3.79µM) of baseline fAAT levels in patients with AATD in this phase 1 study and previously published analyses.30 PK-fAAT time course data from 500 individuals were simulated; 250 individuals with total baseline serum AAT levels of ≤11µM were randomly sampled for further summary.
Results
Patients
Overall, 31 patients were administered INBRX-101. In part 1, 24 patients were enrolled and received 1 intravenous infusion of 10, 40, 80, or 120mg/kg INBRX-101 (each n=6). In part 2, 18 patients (rollover patients from part 1, n=11) received 3 doses of 40, 80, or 120mg/kg INBRX-101 Q3W (each n=6). Per study criteria, patients had documented antigenic serum AAT of <11μM in their medical history. All except 5 patients had a PiZZ genotype, and baseline serum fAAT concentrations were 2.0–17.7μM (Table 1). Approximately half of patients had evidence of emphysema (51.6%), and the median forced expiratory volume in 1 second was 81.0% of predicted. Six patients (19.4%) had previously received pdAAT.
All patients completed dosing, and most completed the study (n=26 [83.9%]; data cutoff, September 26, 2022). Five patients discontinued (consent withdrawal, n=1; inability to complete study visits, n=4 [attributed to the COVID-19 pandemic, n=2]).
Safety
AEs were reported in 28 of 31 patients (90.3%) (Table 2). Most were transient and self-limited, and only 1 grade ≥3 AE was reported (progression of preexisting follicular lymphoma). Fatigue was most common, occurring in 29.0% of patients (treatment related in 7 of 9 patients), followed by upper respiratory tract infections (16.1%). Treatment-related AEs, as determined by the investigator, occurred in 14 patients and were all grade 1 or 2. In 2 patients, serious AEs, both unrelated to the study drug, were reported: the lymphoma described above and sinus tachycardia grade 2, which resolved without treatment. No AEs led to treatment discontinuation or death.
AEs considered to be infusion related occurred in 8 patients. All were grade 1 or 2 and included pruritus, flushing, and urticaria (Supplementary Table 2 in the online supplement). Grade 2 infusion-related AEs were reported in 3 patients but were manageable and resolved on the same day; 2 patients received diphenhydramine and/or famotidine as supportive care. No eosinophil elevations were observed in these patients, and reported event terms suggested that infusion-related events were not due to INBRX-101 hypersensitivity.
Pharmacokinetics and Immunogenicity
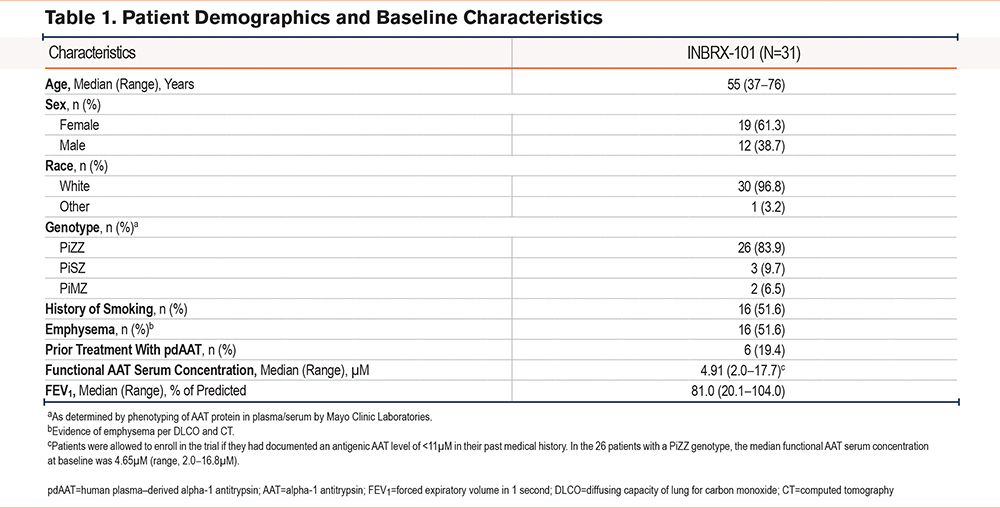
In part 1, INBRX-101 maximal concentration was achieved at or near the end of infusion; dose-related increases in maximal and total INBRX-101 exposure were observed across evaluated doses. The mean terminal elimination half-life was between 12.2 (10mg/kg) and 20.0 days (120mg/kg).
In part 2, similar trends were observed, with dose-proportional increases in plasma area under the INBRX-101 concentration-time curve and maximal concentration (Figure 1B; Supplementary Table 3 in the online supplement). The mean terminal elimination half-life was 15.7 (40mg/kg) to 18.2 days (120mg/kg). Measurable INBRX-101 exposure was observed in all 3 cohorts for ≥28 days after the third dose.
Of the 29 patients evaluable for immunogenicity, ADAs against INBRX-101 were confirmed in 9 patients (treatment emergent, n=6; preexisting, n=3), of whom 1 developed low-titer neutralizing antibodies. Five patients had ADAs (treatment emergent, n=4; preexisting, n=1) reactive to INBRX-101 and AAT; none were neutralizing to AAT. Four patients still had ADAs (treatment emergent, n=2; preexisting, n=2) against INBRX-101 at their final testing.
ADA titers were low and had no meaningful impact on INBRX-101 PK or PD, including in the patient with neutralizing antibodies. PK parameters were comparable between patients regardless of ADA status. ADAs did not result in accelerated mean clearance of INBRX-101 (ADA-positive patients, 0.4mg/(day×µM)/kg; ADA-negative patients, 0.49mg/(day×µM)/kg).
Pharmacodynamics of Serum Functional Alpha-1 Antitrypsin
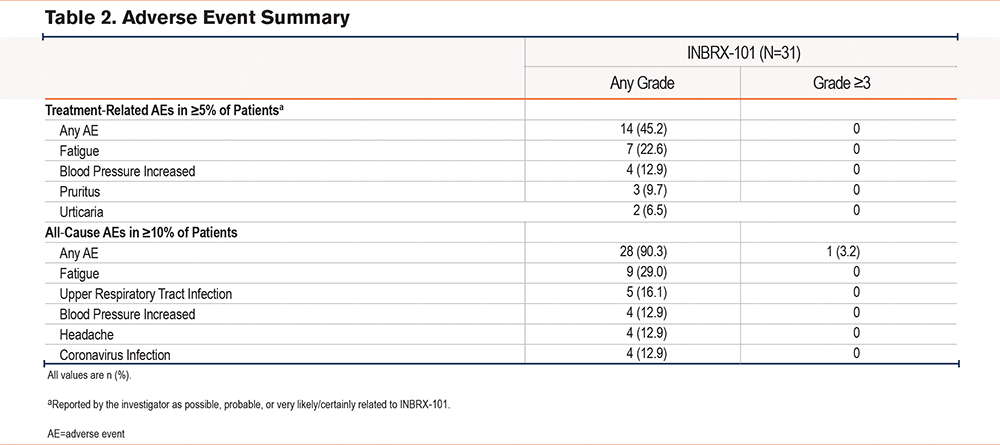
As measured by changes in serum NE inhibitory activity, dose-related increases in serum fAAT were observed in parts 1 and 2 (Figure 1C). Accumulation of serum fAAT corresponded with the long terminal elimination half-life of INBRX-101. For the duration of the dosing interval, 80 and 120mg/kg Q3W INBRX-101 resulted in serum fAAT levels well above the historical target threshold of 11μM antigenic AAT and above 21μM, which was the lower limit of fAAT levels that we estimated previously using our validated fAAT assay in a pilot study of PiMM individuals (range,34 21–54µM). The mean serum fAAT level 21 days after the third dose of INBRX-101 (study day 63) was 35.64µM in the 80-mg/kg cohort and 29.82µM in the 120-mg/kg cohort; mean baseline concentrations were 10.76µM and 8.73µM, respectively (Table 3). The mean serum fAAT level remained >21µM in the 120-mg/kg dose cohort (27.28µM) even 28 days after the third dose (study day 70). The PD profile of serum fAAT in the 26 patients with a PiZZ genotype (Supplementary Figure 2 in the online supplement) was comparable to that observed in the total patient population. ADAs did not impact serum fAAT levels over time (Supplementary Figure 3 in the online supplement).
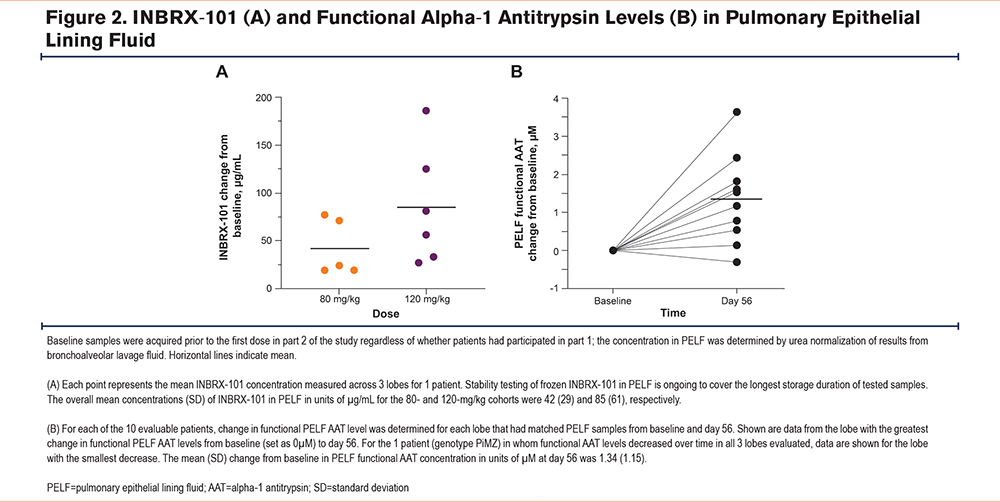
INBRX-101 and Functional Alpha-1 Antitrypsin in Pulmonary Epithelial Lining Fluid
BALF samples were collected before and after dosing in part 2 from 11 patients (80mg/kg, n=5; 120mg/kg, n=6) to determine INBRX-101 and fAAT concentrations in PELF. Using a specific validated mass spectrometry assay, INBRX-101 was detected at day 56 (i.e., 14 days after the third dose) in PELF from all assessed lung lobes of every patient evaluated (Figure 2A). INBRX-101 was also detected in predose samples from part 1 rollover patients, even though ≥84 days had elapsed since their single dose in part 1. Dose-dependent increases of INBRX-101 were seen in PELF, with higher levels on average in patients administered 120 versus 80mg/kg. INBRX-101 remained active in PELF, with fAAT levels in PELF generally higher post baseline, as measured using a validated anti-NE capacity assay (Figure 2B).
Phase 2 Dose Determination
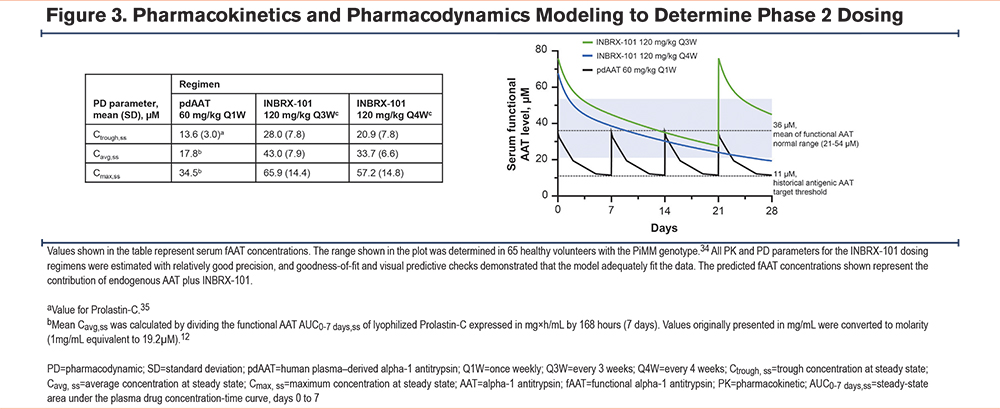
We used PK/PD modeling and simulation to predict INBRX-101 and fAAT levels following steady-state dosing of 120mg/kg INBRX-101 Q3W or every 4 weeks (Q4W). Our modeling projected 120mg/kg Q4W would achieve a mean steady-state average concentration of ≈34µM and a mean steady-state trough concentration of ≈21µM (Figure 3)35 versus ≈43µM and ≈28µM, respectively, with Q3W dosing. Based on these projections and the results from part 2, both dosing schedules will be evaluated in a pivotal phase 2 trial.
Discussion
This study evaluated the safety of a novel recombinant AAT, INBRX-101, in patients with AATD. INBRX-101 was designed to overcome the limitations of pdAAT, with an improved PK profile, including a longer half-life. In part 2 of this phase 1 study, INBRX-101 exhibited a mean half-life ≈2.5 to >3 times of that documented for pdAAT (16–18 days versus 5–7 days12-16). The greater exposure to INBRX-101 corresponded with dose-dependent increases in serum fAAT levels. As INBRX-101 contains 2 AAT molecules, the serum concentration of fAAT was approximately double that of INBRX-101, with some deviation from a 2:1 ratio expected due to the contributions of endogenous fAAT. Consistent with the prolonged half-life of INBRX-101, accumulation of INBRX-101, and consequently serum functional AAT activity, was observed following each dose. The mean average concentration of serum fAAT over the 21-day interval following the third dose of INBRX-101 in the 80- and 120-mg/kg cohorts (39.9µM and 43.3µM, respectively) was well above the 21µM lower limit of serum fAAT levels that was observed in PiMM individuals. In patients who received 120mg/kg INBRX-101, mean serum fAAT levels continued to be >21µM at 21 and 28 days after the third dose (29.8µM and 27.3µM, respectively). In contrast, for pdAAT, a mean steady-state trough concentration of 16.7µM to 16.9µM has been reported for total antigenic serum AAT, of which only a portion is functional.12
Importantly, INBRX-101 penetrated the lungs and was detected in all 3 lobes assessed in each of the 11 evaluable patients. INBRX-101 was detected prior to dosing in part 2 only in patients dosed during part 1, confirming our assay’s specificity. Mean INBRX-101 PELF concentrations were greater in patients who received 120mg/kg INBRX-101, and in general, fAAT levels increased following INBRX-101 administration. The latter suggests that INBRX-101 contributes to the greater anti-NE activity in PELF, but extending these analyses to a larger patient population is the subject of future research.
INBRX-101 was generally well tolerated, with only 1 grade ≥3 AE reported (progression of preexisting follicular lymphoma). This was expected given that pdAAT has not been associated with overt toxicity, even at doses of >60mg/kg,32,36-40 and serum AAT levels can become considerably elevated as part of the body’s natural immune response.41,42 All patients completed dosing; 5 discontinued before completing all study visits for reasons unrelated to INBRX-101. As INBRX-101 is a recombinant protein, infusion-related AEs were of special interest. Such AEs were reported in 8 patients, were all grade 1 or 2, resolved on the same or next day with no to minimal supportive care, and appeared to be a function of infusion volume versus hypersensitivity to INBRX-101. The incidence of ADAs against INBRX-101 was low overall, occurring in 9 of 29 evaluable patients. ADA titers were low and had no observed impact on serum INBRX-101 PK or fAAT PD.
pdAAT has been the standard of care for patients with severe AATD since the 1980s; however, it is associated with several limitations. As a consequence of its relatively short half-life,12,13 patients need weekly infusions, which can negatively impact quality of life and affect treatment adherence.43 Furthermore, the approved regimen of pdAAT 60mg/kg once weekly strives for serum AAT levels above a target threshold of 11µM AAT,2,12,13,21 which is approximately half of the lower limit (20µM) observed in PiMM individuals.22 pdAAT 60mg/kg fails to maintain serum antigenic AAT levels of >20µM beyond the first 3−5 days of the weekly dosing period.19,21,32 Although historically considered the minimal protective concentration of serum AAT, 11µM as the target threshold of treatment has been increasingly questioned,23-28 with recent evidence of greater benefit at higher levels.30-32 Additionally, as a human blood product, pdAAT sourcing is vulnerable to disruptions of the human blood supply, as experienced during the COVID-19 pandemic.44
Fc fusion protein technology, which has been successfully used in therapies for other disease areas,45 enables the extended half-life observed for INBRX-101. Unlike pdAAT, INBRX-101 allows patients to sustain serum antigenic AAT levels of >20µM for 3−4 weeks between doses as opposed to the 3–5 days observed for pdAAT,19,21,32 potentially leading to better outcomes. The longer half-life of INBRX-101 also results in less frequent dosing and thus, less patient burden, which may improve treatment compliance. Additionally, as INBRX-101 is a recombinant protein, manufacturing is scalable, resulting in a predictable and reliable supply independent of human blood donations. This could more readily allow for dose-optimization studies or evaluations in patients with more moderate AATD without the accompanying ethical concerns of diverting limited supply.2,26,27,29 Furthermore, INBRX-101 is free of the inherent risk for transmission of bloodborne pathogens.
Overall, 80 and 120mg/kg INBRX-101 Q3W led to serum fAAT levels of >21µM for the entire dosing interval, with a safety profile similar to that of pdAAT. These levels of fAAT were achieved by administering INBRX-101 Q3W instead of weekly, as done with pdAAT.2,10,11 Notably, 120mg/kg Q3W led to fAAT serum levels of >21µM even 4 weeks after the last dose of INBRX-101. Furthermore, INBRX-101 was shown to penetrate the lungs, which suggested activity in the target organ. PK/PD modeling using data from this trial predicts that 120 mg/kg INBRX-101 dosed on a less frequent Q4W schedule would still maintain a mean steady-state trough concentration of serum fAAT at 21µM.
The findings of this trial support further evaluation of 120mg/kg INBRX-101 Q3W and Q4W as possible treatments for patients with AATD. A double-blind phase 2 trial assessing INBRX-101 in patients with AATD at a dose of 120mg/kg Q3W or Q4W is underway (ElevAATe, NCT05856331). This randomized trial will evaluate INBRX-101 versus pdAAT, with a primary endpoint of change in serum fAAT concentration from baseline to trough at steady state. Patients eligible for ElevAATe will have centrally confirmed serum antigenic AAT concentrations of <11µM at screening and genotypes associated with more severe deficiency (e.g., PiZZ and PiZ/Null). Patients with PiSZ, PiMS, or PiMZ will be excluded. In addition to a more homogenous population, ElevAATe will have a greater number of patients from whom BALF samples are collected, allowing further evaluation of the connection between INBRX-101 and functional AAT concentrations in PELF.
Acknowledgements
Author contributions: MLB, BTK, HWF, RM, AC, CLC, CDB, MAC, JEL, and AV were responsible for the investigation, supervision, resources, writing–original draft, writing–review, and editing. EB, SB, TB, CV, VA, JK, and BPE were responsible for the conceptualization, data curation, formal analysis, methodology, validation, visualization, writing–original draft, writing–review, and editing. All authors agreed to the submission of the manuscript for publication.
Data sharing statement: The protocol for and the data generated and/or analyzed in the current study are not publicly available but are available upon reasonable request after the entire study is completed. Sharing of data is subject to protection of patient privacy and respect for the patient’s informed consent. For more information on the process or to submit a request, contact clinicaltrials@inhibrx.com.
We thank the study participants and their families; all the INBRX-101 phase 1 trial investigators; the Inhibrx, Inc., research team; Daniel Tompkins, Tina Tham, and Karla Ramirez who were instrumental in enrollment at the University of California- Davis; and all other personnel involved in the study for their participation and contributions. We also thank Jenna M. Gaska, PhD, of Nucleus Global, an Inizio company, for medical writing assistance.
Declaration of Interests
MLB reports a relationship with the University of Florida College of Medicine that includes nonfinancial support. BTK reports a relationship with Takeda that includes consulting or advisory and speaking and lecture fees; a relationship with Boehringer Ingelheim that includes consulting or advisory fees; a relationship with Grifols that includes speaking and lecture fees; a relationship with Inhibrx, Inc., that includes consulting or advisory fees and travel reimbursement; and a relationship with the California Thoracic Society that includes nonfinancial support. HWF reports financial support provided by Hannibal Clinic Operations, LLC, and a relationship with Hannibal Clinic Operations, LLC, that includes employment. RM reports a relationship with Intellia Therapeutics, Inc., that includes consulting or advisory fees. CDB is an investigator for the ElevAATe trial (NCT05856331) that is sponsored by Inhibrx, Inc. MAC reports a relationship with Inhibrx, Inc., that includes consulting or advisory fees. EB reports a relationship with Inhibrx, Inc., that includes employment and equity or stocks. SB reports a relationship with Inhibrx, Inc., that includes employment and equity or stocks and has a patent titled “Effective Dosage of Recombinant Serpin-Fc Fusion Protein for Use in a Method of Treating AAT Deficiency in a Subject” pending to Inhibrx, Inc. TB reports a relationship with Inhibrx, Inc., that includes employment and equity or stocks. CV reports a relationship with Inhibrx, Inc., that includes employment and equity or stocks. VA reports a relationship with Inhibrx, Inc., that includes employment and equity or stocks. JK reports a relationship with Inhibrx, Inc., that includes employment and equity or stocks and reports a patent titled “Effective Dosage of Recombinant Serpin-Fc Fusion Protein for Use in a Method of Treating AAT Deficiency in a Subject” pending to Inhibrx, Inc. BE reports a relationship with Inhibrx, Inc., that includes employment and equity or stocks and reports patent #10,400,029 “Serpin Fusion Polypeptides and Methods of Use Thereof” issued to Inhibrx, Inc. AC, CLC, JEL, and AGV have nothing to disclose.