Running Head: CXCL12 From IL-17A-Activated Fibroblasts in COPD
Funding Support: This work was supported by the National Natural Science Foundation of China (Grant No. 81800040) and funded by the Project of NINGBO Leading Medical & Health Discipline (Project Number: 2022-B19).
Date of Acceptance: July 3, 2024 | Publication Online Date: July 9, 2024
Abbreviations: α-SMA=alpha-smooth muscle actin; AMD3100=CXCR4 antagonist; BALF=bronchoalveolar lavage fluid; cDNA=complimentary DNA; COPD=chronic obstructive pulmonary disease; CS=cigarette smoke; CXCL12=C-X-C motif chemokine ligand 12; CXCR4=C-X-C motif chemokine receptor 4; DAPI=4',6-diamidino-2-phenylindole stain; DMEM=Dulbecco’s Modified Eagle Medium; E-cadherin=epithelial-cadherin; ELISA=enzyme-linked immunosorbent assay; EMT=epithelial-mesenchymal transition; ERK=extracellular signal-regulated kinase; FBS=fetal bovine serum; GOLD=Global initiative for chronic Obstructive Lung Disease; HBE=human bronchial epithelial; HE=hematoxylin and eosin; HLF=human lung fibroblasts; HPCs=hematopoietic progenitor cells; IL-17A=interleukin-17A; IgG=immunoglobin G; MAPK=mitogen-activated protein kinase; mRNA=messenger RNA; p-ERK=phosphorylated extracellular signal-regulated kinase; PBS=phosphate-buffered saline; PCR=polymerase chain reaction; rIL-17A=recombinant interleukin 17A; RT-qPCR=reverse transcription quantitative polymerase chain reaction; SD=standard deviation; Th=T helper cell
Citation: Chen X, Chen L, Chen G, et al. Interleukin-17A promotes airway remodeling in chronic obstructive pulmonary disease by activating C-X-C motif chemokine ligand 12 secreted by lung fibroblasts. Chronic Obstr Pulm Dis. 2024; 11(5): 482-495. doi: http://doi.org/10.15326/jcopdf.2024.0495
Online Supplemental Material: Read Online Supplemental Material (119KB)
Background
Chronic obstructive pulmonary disease (COPD), a common chronic airway inflammatory disease, is characterized by a progressive and irreversible decline in pulmonary function. According to the 2023 Global initiative for chronic Obstructive Lung Disease (GOLD) report,1 COPD is a leading cause of death worldwide. According to the Global Burden of Disease database,2 its incidence rate was 0.04% in the total global population and the mortality rate was 5.8%. Macrophages as well as CD4>+ and CD8+ T cells, along with their secreted cytokines, are involved in the development of COPD.3 Cigarette smoke (CS) exposure is a risk factor for COPD, and continuous CS exposure leads to COPD progression and changes to the structure of the airways, which is termed airway remodeling. In patients with COPD, airway remodeling mainly occurs in the peripheral airways and is characterized by airway wall thickening and fibrosis, goblet cell hyperplasia, mucus hypersecretion, and smooth muscle hyperplasia and hypertrophy.4 Features of airway wall fibrosis include increased deposition of extracellular matrix proteins, particularly collagens I and III, fibronectin, and proteoglycans.5
Epithelial-mesenchymal transition (EMT) may initiate airway remodeling in COPD. EMT refers to the biological process in which the epithelial cells acquire properties characteristic of mesenchymal stem cells, which are involved in embryonic development, but also in disease progression, such as cancer metastasis and various fibrotic diseases.6 It is characterized by the loss of epithelial cell markers, such as cytokeratin, tight junction proteins, and E-cadherin (epithelial-cadherin), as well as by the acquisition of mesenchymal cell markers, such as vimentin and alpha-smooth muscle actin (α-SMA). In CS-induced COPD, epithelial cells transform into fibroblasts through EMT, leading to periairway fibrosis following their activation.7 Therefore, fibroblast activation is being investigated as one of the mechanisms responsible for airway remodeling in COPD.
Considering that interleukin-17A (IL-17A) is mainly produced by T helper (Th) 17 cells and is present during COPD stabilization and acute exacerbation,8 Dessalle et al9 suggested that IL-17A could promote fibroblast activation in the lungs. Then, inhibition of IL-17A production could prevent airway remodeling by controlling airway inflammation and regulating fibroblast activation in COPD. However, the effect of IL-17A on lung fibroblasts remains unknown.
C-X-C motif chemokine ligand 12 (CXCL12), also known as stromal cell-derived factor 1, is a chemokine secreted during fibroblast activation. By specifically binding to its receptor, C-X-C motif chemokine receptor 4 (CXCR4), it triggers the chemotaxis of T lymphocytes and monocytes, which are involved in Th2-type airway anaphylaxis.10 Its concentration in the bronchoalveolar lavage fluid (BALF) was significantly higher in patients with asthma than in healthy controls.11 CXCL12 is also involved in chronic lung inflammation.12 Following the use of the CXCR4 antagonist AMD3100 in a mouse model13 of ovalbumin-induced asthma, the levels of IL-17A in lung tissues were significantly decreased, accompanied by a simultaneous decrease in the Th17 cell-type immune responses.14 Dupin et al15 found elevated levels of CXCR4-expressing fibrocytes in the peripheral blood and increased migration of these fibrocytes to lung tissues in patients with an acute exacerbation of COPD, which was involved in the development of chronic airway inflammation in COPD. Accordingly, the relationship between CXCL12/CXCR4 and airway remodeling in COPD should be further explored.
The aim of the present study was to investigate the ability of IL-17A to activate lung fibroblasts and upregulate CXCL12 secretion, which would in turn lead to airway remodeling in COPD. The possible mechanisms underlying the involvement of IL-17A in airway remodeling were also explored.
Methods
Animals
Male BALB/c mice (6–8 weeks old) were purchased from the Laboratory Animal Center of the Zhejiang Academy of Medical Sciences. The animals were housed in isolated ventilated cages (5 mice per cage) at a controlled temperature (20°–23°C), under a 12-hour light/dark cycle with 45%–65% humidity. Standard laboratory chow and water were provided ad libitum. The euthanasia procedure for mice was performed via an intraperitoneal injection of 50mg/kg sodium barbiturate. The study was approved by the Ethics Committee of Ningbo University Affiliated People’s Hospital and the Experimental Animal Ethics Committee of Ningbo University. All animal experimental protocols were approved by the Experimental Animal Ethics Committee of Ningbo University, and all experimental procedures followed the Guidelines of the Care and Use of Laboratory Animals issued by the Chinese Council on Animal Research. All methods were carried out in accordance with the relevant guidelines and regulations. Written informed consent to participate in the study was obtained from all patients. All animals received humane care in accordance with the Guide for the Care and Use of Laboratory Animals of Ningbo University. The study was performed in accordance with the ARRIVE guidelines, and the work conformed to the policies in the 1996 Guide for the Care and Use of Laboratory Animals.
Cell Culture
Lung tissues were removed from healthy BALB/c mice, and they were quickly washed with phosphate-buffered saline (PBS) containing antibiotics (100U/mL penicillin and 100µg/mL streptomycin) and minced to 1–2mm3 pieces using sterile tools. The tissue fragments were added to Dulbecco’s Modified Eagle Medium (DMEM; 10mL) containing fetal bovine serum (FBS; 10%) and penicillin-streptomycin (1%), then they were evenly distributed on a 10cm plate. Subsequently, the lung tissue fragments were incubated at 37°C with 5% carbon dioxide (CO2) and saturated humidity for almost 2 weeks until the fibroblasts had exited the tissue and attached to the plate. Media was replaced every 2 days. When the fibroblast density reached approximately 80%, the fragments were removed and the first passage was performed. Mouse primary lung fibroblasts were used for experiments after the third passage. Human lung fibroblasts were isolated from fresh human lung tissues using the same culture processes and conditions.16,17 We obtained the lung tissue from patients who underwent pulmonary nodule resection with the help of a thoracic surgeon, and the lung tissue was obtained from a location, whose distance was greater than 5cm from the lesion. Patient characteristics are described in Supplementary Table 1 in the online supplement. Recombinant (r)IL-17A (421-ML-025/ 317-ILB-050, R & D Systems) was used to activate lung fibroblasts at concentrations of 50, 100, and 200ng/ml. All experimental protocols were approved by the Ethics Committee of Ningbo University Affiliated People’s Hospital, and the methods were carried out in accordance with the relevant guidelines and regulations. Written informed consent to participate in the study was obtained from all patients.
The human bronchial epithelial (HBE) cell line was purchased from American Type Culture Collection (Manassas, Virginia), and it was maintained in DMEM supplemented with 10% FBS and antibiotics (penicillin 100U/mL, streptomycin 100μg/mL) in a humidified atmosphere with 5% CO2 at 37°C.
Establishment of a Mouse Model of COPD and Collection of Bronchoalveolar Lavage Fluid
A total of 60 male mice were randomly divided into 4 groups (n = 15 per group), comprising the control, COPD, COPD + anti-IL-17A antibody, and COPD + AMD3100 groups. Control mice were housed in a standard environment, while the remaining mice (n = 45) were exposed to CS generated from research-grade cigarettes (3R4F; University of Kentucky, Lexington, Kentucky) in a square plastic box lasting 1 hour per day, 7 days per week, for a total of 24 weeks. To investigate the effects of IL-17A and CXCL12 on COPD, all 60 mice were treated with vehicles on day 28 of CS exposure. Mice in the COPD + anti-IL-17A antibody group were treated with 50µg (0.2mL) of an anti-IL-17A antibody via an intraperitoneal injection for 2 days per week. Mice in the COPD + AMD3100 group were treated with 5mg/kg (0.2mL) of AMD3100 via an intraperitoneal injection for 5 days per week. Mice in the control and COPD groups were treated with 0.2mL of PBS via an intraperitoneal injection for 5 days per week. The doses were determined based on a previous study18 and preliminary experiments. Mice were sacrificed by intraperitoneal injection of 50mg/kg barbiturate sodium in 24 hours after that last CS challenge. Unilateral lung was lavaged 3 times with 1ml of Hanks’ Balanced Salt Solution (Sangon Biotech, Shanghai, China).
Enzyme-Linked Immunosorbent Assay
CXCL12 levels were measured in the supernatant from cell cultures and BALF by using an enzyme-linked immunosorbent assay (ELISA) kit (Elabscience Biotechnology, Wuhan, China), according to the manufacturer’s instructions.
Histological Assessment of Lung Tissues
Lung tissues of mice were excised, fixed in paraformaldehyde (4%), embedded in paraffin, and cut into sections (4µm). The sections were stained with hematoxylin and eosin following standard protocols. The mean linear intercept was used to measure the alveolar size in the mouse tissue sections to evaluate the degree of emphysema, as described in the literature.19 Briefly, a 10x objective slice image was imported into Image-ProPlus 6.0, and a cross line of length L was placed in the center of each picture and the number N of intersections between the alveolar septa and it was automatically identified by the software. We defined L/N as mean linear intercept.
Immunohistochemical Staining
Immunohistochemical staining was performed using the avidin–biotin–peroxidase complex method. Briefly, 4% paraformaldehyde-fixed and paraffin-embedded murine lung tissue samples were cut into sections (4 µm). The sections were first heated at 56°C for 30 minutes. Subsequently, deparaffinization with xylene and rehydration with an ethanol gradient was performed. The slides were incubated in 3% hydrogen peroxide to block the endogenous peroxidase activity. For antigen retrieval, the sections were boiled for 10 minutes in Antigen Unmasking Solution (Vector Laboratories, Newark, California). Primary antibodies against E-cadherin (1:200 dilution, #14472, CTS), α-SMA (1:200 dilution, NBP2-78836, Novus Biologicals, Centennial, Colorado), collagen I (1:200 dilution, ab6308, Abcam, Cambridge, United Kingdom), vimentin (1:200 dilution, ab20346, Abcam), or CXCL12 (1:100 dilution, ab9797, Abcam) were used to label the sections at 4°C overnight. Anti-mouse (dilution 1:200, BA9400, Vector Laboratories) or anti-rabbit (dilution 1:200, BA1000, Vector Laboratories) immunoglobin G (IgG) antibodies were applied using the ABC Peroxidase Standard Staining Kit (PK-4000, Vector Laboratories) at 25°C for 1 hour. Each slide was allowed to react with the 3,3'-diaminobenzidine Substrate Kit (SK-4100, Vector Laboratories) for 10–20 seconds, and then, it was counterstained with hematoxylin for 5 minutes. For immunohistochemical scoring, 2 pathologists randomly selected 5 areas in each slice under a microscope (Axio Lab.A1; Zeiss AG, Germany) with a 10X objective according to the distribution (0 for < 5%, 1 for 5% to 25%, 2 for 26% to 50%, 3 for 51% to 75%, and 4 for 76% to 100%) and pigmentation depth (0 for colorless, 1 for yellowish, 2 for brownish-yellow, and 3 for tan) of target protein staining. The score product of the distribution and coloration was the final fraction of the field.
For the immunofluorescence assay, lung fibroblasts grown on slides were fixed with paraformaldehyde (4%). After blocking with normal nonimmune goat serum for 20 minutes, the fibroblast slides were incubated with antibodies against vimentin (1:200 dilution, ab20346, Abcam), collagen I (1:200 dilution, ab6308, Abcam), or CXCL12 (1:500 dilution, ab9797, Abcam), and then stained with the appropriate fluorescein isothiocyanate-conjugated anti-mouse IgG antibody (1:200 dilution, ab6785, Abcam) or Alexa Fluor® 488-conjugated anti-rabbit IgG antibody (1:200 dilution, A27039, Invitrogen, Waltham, Massachusetts).
Reverse Transcription Quantitative Polymerase Chain Reaction
Total RNA was isolated using the TRIzol Reagent (Invitrogen, Carlsbad, California) according to the manufacturer’s instructions. RNA was reverse-transcribed into complementary (c)DNA using oligo(dT) primers and a reverse transcriptase (TAKARA, Shiga, Japan). cDNA was used for subsequent reverse transcription quantitative polymerase chain reaction (RT-qPCR) analysis with the Fast SYBR Green Master Mix (Applied Biosystems, Foster City, California). Each reaction was performed on a Mastercycler PCR machine (Applied Biosystems). Primer sequences are listed in Table 1. The relative expression of the gene of interest was normalized to the housekeeping gene β-actin. Data were analyzed using the comparative CT method. Specificities of the resulting PCR products were confirmed using melting curves.
Western Blotting
Fibroblasts and mouse lung tissues were lysed using radioimmunoprecipitation assay cell lysis buffer in the presence of a protease inhibitor (Beyotime, Shanghai, China) and a phosphatase inhibitor cocktail (Sangon Biotech). The total protein concentration was determined using a bicinchoninic acid Protein Assay Kit (Beyotime). Proteins were separated by sodium dodecyl-sulfate polyacrylamide gel electrophoresis on a 10% gel and transferred to poly (vinylidene fluoride) membranes (Bio-Rad, Hercules, California). After blocking with skimmed milk, the membranes were incubated with primary antibodies against E-cadherin (#14472, CTS), α-SMA (NBP2-78836, Novus Biologicals), vimentin (ab20346, Abcam), ERK1/2 (4695, Cell Signaling Technology, Danvers, Massachusetts), phosphorylated-ERK (p-ERK)1/2(4370, Cell Signaling Technology), and β-actin (HC201-01, TransGen Biotech, Beijing, China) diluted at 1:1000 in PBS with Tween 20 overnight at 4°C. The next day, membranes were incubated with the appropriate horseradish peroxidase-conjugated secondary antibody (anti-mouse IgG, 1:5000, TransGen Biotech, or anti-rabbit IgG, 1:5000, Abcam) for 2 hours at 25°C. Finally, a Qinxiang detection system captured the protein band images using an enhanced chemiluminescence reagent (Bio-Rad).
Statistical Analysis
The mean linear intercept of murine lung tissue was measured using Image-Pro Plus 6.0 (Media Cybernetics, Rockville, Maryland). Data were expressed as mean ± standard deviation. All statistical analyses were performed using GraphPad Prism 7.0 (GraphPad, Boston, Massachusetts). The results were analyzed using one-way analysis of variance and Tukey’s test or Bonferroni’s test for the post-hoc test. A p-value < 0.05 was considered statistically significant.
Results
Interleukin-17A Activated Primary Mouse Lung Fibroblasts and Increased C-X-C Motif Chemokine Ligand 12 Secretion
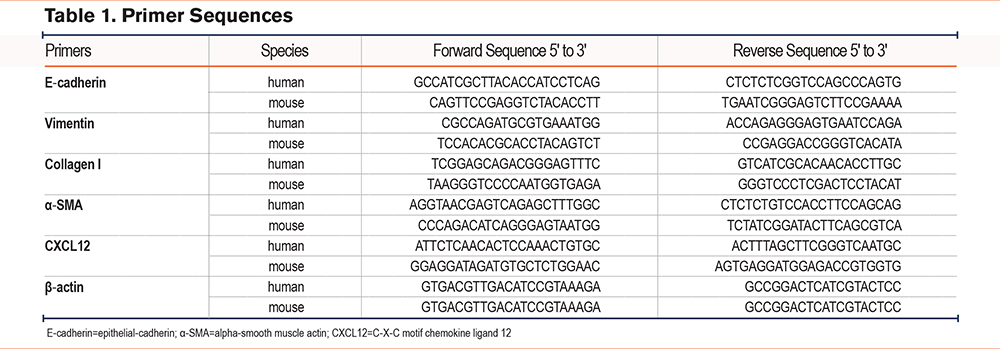
Primary lung fibroblasts were successfully isolated from murine lung tissues and stably passaged 3 times. To confirm the isolated cell type, immunofluorescence staining was performed and the cells were found to express vimentin, consistent with lung fibroblasts (Figure 1A).
To examine the effect of IL-17A on primary mouse lung fibroblasts, cells were treated with either 50, 100, or 200ng/mL of rIL-17A, and then the markers of fibroblast activation were examined. RT-qPCR showed that the messenger (m)RNA expression levels of vimentin, α-SMA, and collagen I were increased after continuous stimulation with rIL-17A for 24 hours compared to those in the control group. The largest differences in expression were seen following stimulation with 100ng/mL of rIL-17A (Figure 1B-D). Additionally, western blotting was performed to examine the protein expression levels of vimentin and α-SMA. Consistent with the RT-qPCR results, the protein expression levels of vimentin and α-SMA were increased in response to rIL-17A stimulation, and the most pronounced increase in levels was observed at a concentration of 100ng/mL (Figure 1E-G). To determine whether mouse lung fibroblasts secreted CXCL12 in response to rIL-17A stimulation, cells and supernatants were collected after 24 hours of rIL-17A exposure. Immunofluorescence staining using a CXCL12-specific antibody revealed that CXCL12 expression was significantly increased with rIL-17A treatment, particularly at a concentration of 100ng/mL (Figure 1H). RT-qPCR and immunofluorescence staining demonstrated upregulation of CXCL12 expression at the mRNA level (Figure 1I) within cells, and an ELISA confirmed CXCL12 secretion (Figure 1J). The optimal concentration of rIL-17A was 100ng/mL. These data suggested that treatment with rIL-17A could activate mouse lung fibroblasts and enhance the secretion of CXCL12.
Challenge With Cigarette Smoke-Induced Emphysema in Mouse Lung Tissues and Increased C-X-C Motif Chemokine Ligand 12 Expression
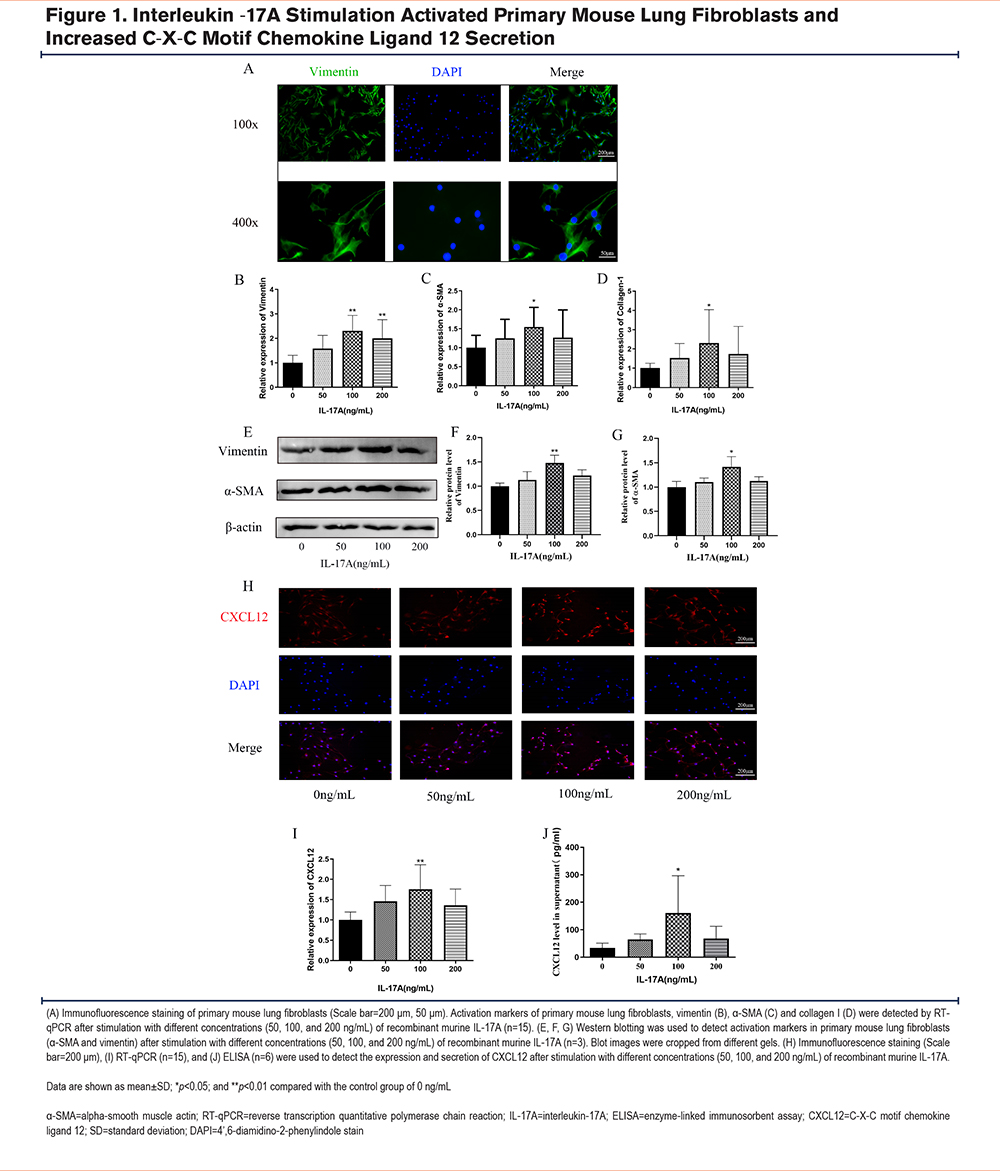
After 24 weeks of CS challenge and treatment with an anti-IL-17A antibody or AMD3100, lung tissues were harvested from mice. Hematoxylin and eosin staining showed that compared to the control group, lung tissues in the CS-attack group had significant alveolar space enlargement, severe alveolar wall thinning, rupture of the alveolar septa, and fusion of alveolar spaces (Figure 2A). The mean linear intercept revealed that the alveolar cavities were larger in the COPD model group than in the control group (Figure 2B, p<0.001). These impairments were attenuated by both the anti-IL-17A antibody and AMD3100 treatments (Figure 2A, B). To assess CXCL12 expression in the 4 experimental groups, BALF was collected at the end of the 24-week treatment period and an ELISA was performed to detect CXCL12. The levels of CXCL12 were significantly higher in the COPD mouse model group compared to the control group (Figure 2E, p<0.01). Additionally, treatment with either the anti-IL-17A antibody (Figure 2E, p<0.05) or AMD3100 (Figure 2E, p<0.05) downregulated CXCL12 secretion in the BALF compared to that in the COPD group. Immunohistochemical results of CXCL12 revealed significantly higher levels of CXCL12 in lung tissues of the COPD mouse model group compared to the control group (Figure 2C, D, p<0.01). CXCL12 levels in the COPD + anti-IL-17A antibody and COPD + AMD3100 groups were slightly higher compared to those in the control group, and they were significantly lower compared to those in the COPD mouse model group (Figure 2C, D; p<0.05 for all). The CXCL12 content in murine lung tissues was further assessed using RT-qPCR. The RNA expression of CXCL12 was higher in the COPD group than in the control group (Figure 2F, p<0.01). The RNA expression levels of CXCL12 in the COPD + anti-IL-17A antibody and COPD + AMD3100 groups were slightly higher compared to those in the control group, but they were significantly lower compared to those in the COPD group (Figure 2F, p<0.05 for all).
Anti-Interleukin-17A or AMD3100 Ameliorated Epithelial-Mesenchymal Transition in Mouse Lung Tissues Under Cigarette Smoke Challenge
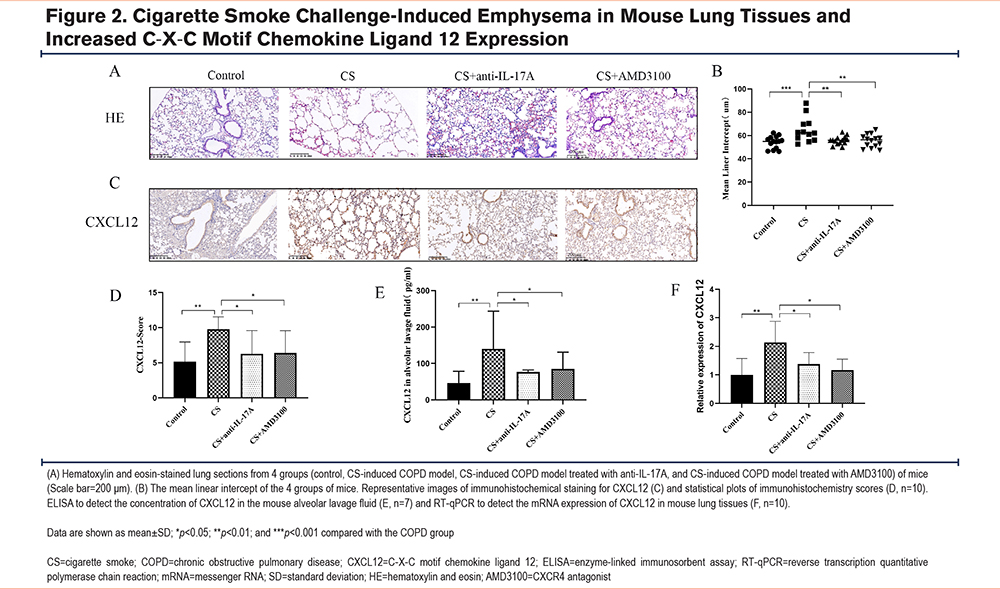
To investigate the effects of long-term CS challenge and anti-IL-17A antibody and AMD3100 treatments on EMT, mouse lung tissues were collected at the end of the 24-week treatment period. The protein expression levels of EMT-related markers, including E-cadherin, vimentin, collagen I, and α-SMA, were examined in each group using immunohistochemistry. In the COPD mouse model, E-cadherin expression was decreased, (Figures 3A and a) while the expression of vimentin (Figures 3B and b), collagen I (Figures 3C and c), and α-SMA (Figures 3D and d) was increased. However, treatment with either the anti-IL-17A antibody or AMD3100 reversed the CS-induced changes in EMT-related marker expression (Figure 3A–D, Figure 3a-d). CS decreased the mRNA expression of E-cadherin (Figure 3E), and it increased the mRNA expression of vimentin (Figure 3F), collagen I (Figure 3G), and α-SMA (Figure 3H). Figure 3I-L shows the EMT-related markers in each group, and the trends were consistent with the immunohistochemistry and RT-qPCR results. Taken together, treatment with either the anti-IL-17A antibody or AMD3100 was able to reverse EMT in the mouse model of CS-induced COPD.
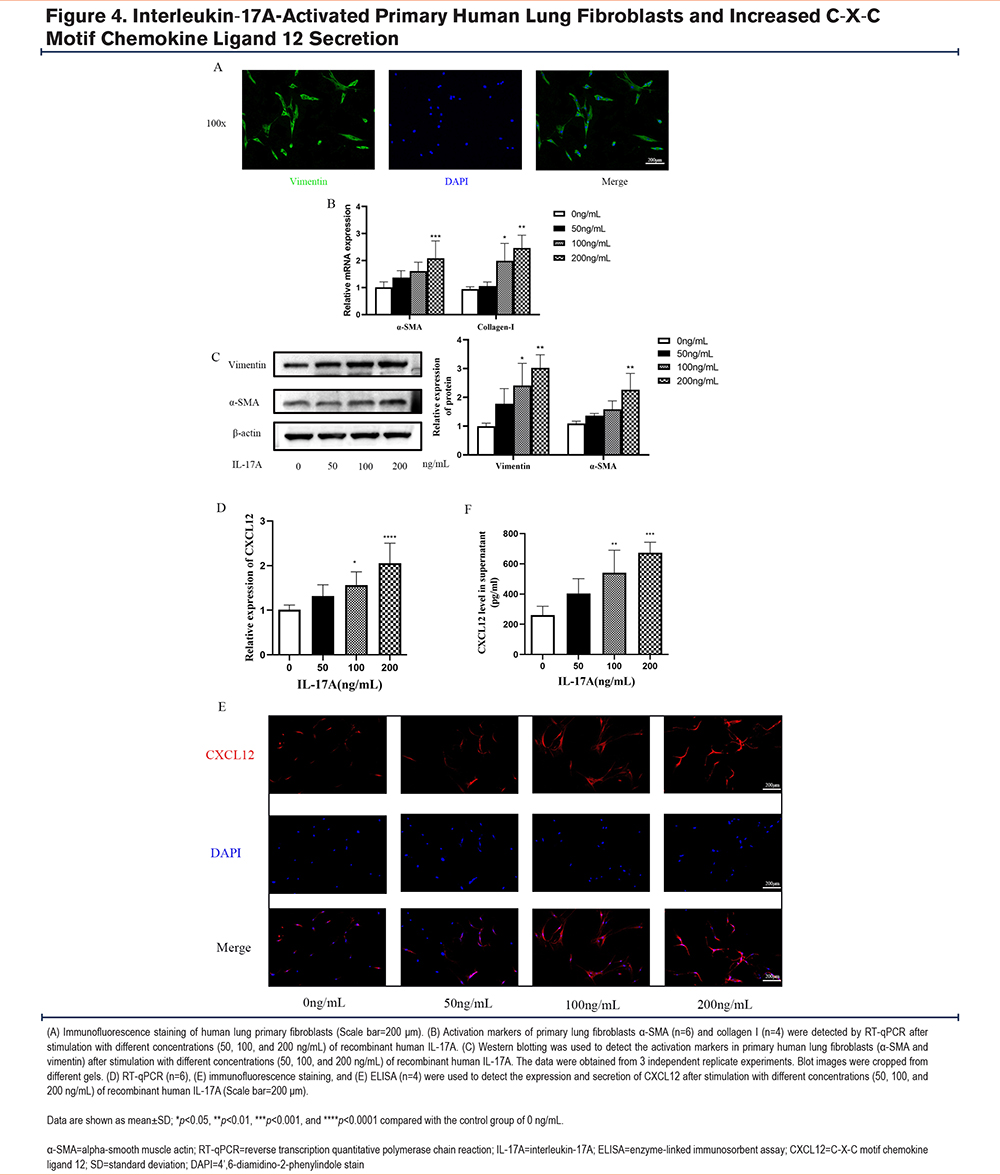
Interleukin-17A Stimulation Caused Activation of Human Lung Primary Fibroblasts and Increased C-X-C Motif Chemokine Ligand 12 Secretion
Primary human fibroblasts were isolated from the human lung tissue and were confirmed by immunofluorescence staining (Figure 4A). Following stimulation with human rIL-17A, markers of fibroblast activation were measured by RT-qPCR and western blotting. Levels of human fibroblast activation were increased in a concentration-dependent manner (Figure 4B, C). RT-qPCR and immunofluorescence staining showed a trend of higher CXCL12 expression in fibroblasts following rIL-17A treatment (Figure 4D-E). An ELISA was used to measure the level of CXCL12 in the cell supernatant. Compared to that in the control group, human rIL-17A increased the production of CXCL12 in a concentration-dependent manner (Figure 4F). Therefore, among the concentrations tested, IL-17A had the greatest effect on human fibroblast activation at a concentration of 200ng/mL.
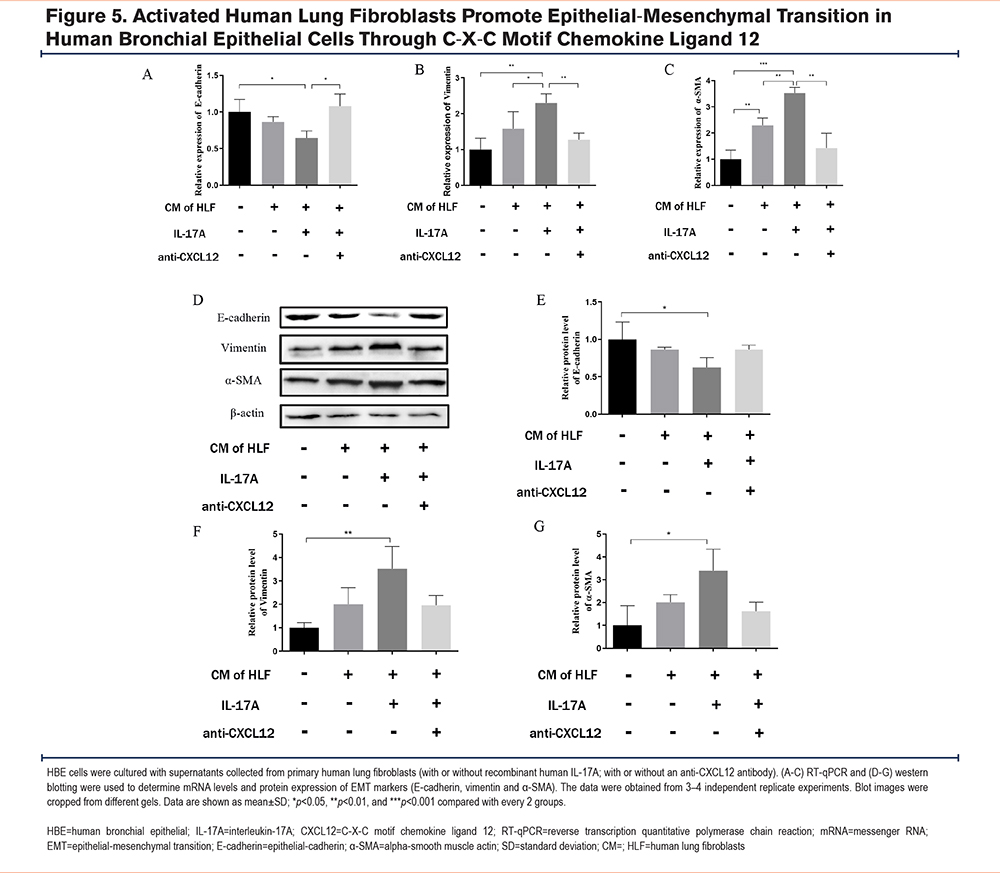
Activated Human Lung Fibroblasts Promoted Epithelial-Mesenchymal Transition in Human Bronchial Epithelial Cells Through C-X-C Motif Chemokine Ligand 12
After determining that the effective concentration of IL-17A for stimulation was 200ng/mL, cell supernatants were collected from untreated human lung fibroblasts and fibroblasts treated with 200ng/mL of rIL-17A. These cell supernatants were added to HBE cells for 48 hours. Total RNA and protein of HBE cells were extracted to detect the markers of EMT. Following culture of HBE cells with the rIL-17A-stimulated fibroblast supernatant, the expression of E-cadherin (Figure 5A, D, E) was decreased, and the expression of vimentin (Figure 5B, D, F) and α-SMA (Figure 5C, D, G) was increased, suggestive of EMT. When an anti-CXCL12 antibody was added to the fibroblast supernatant to neutralize CXCL12, the EMT process initiated in the HBE cells was significantly reversed.
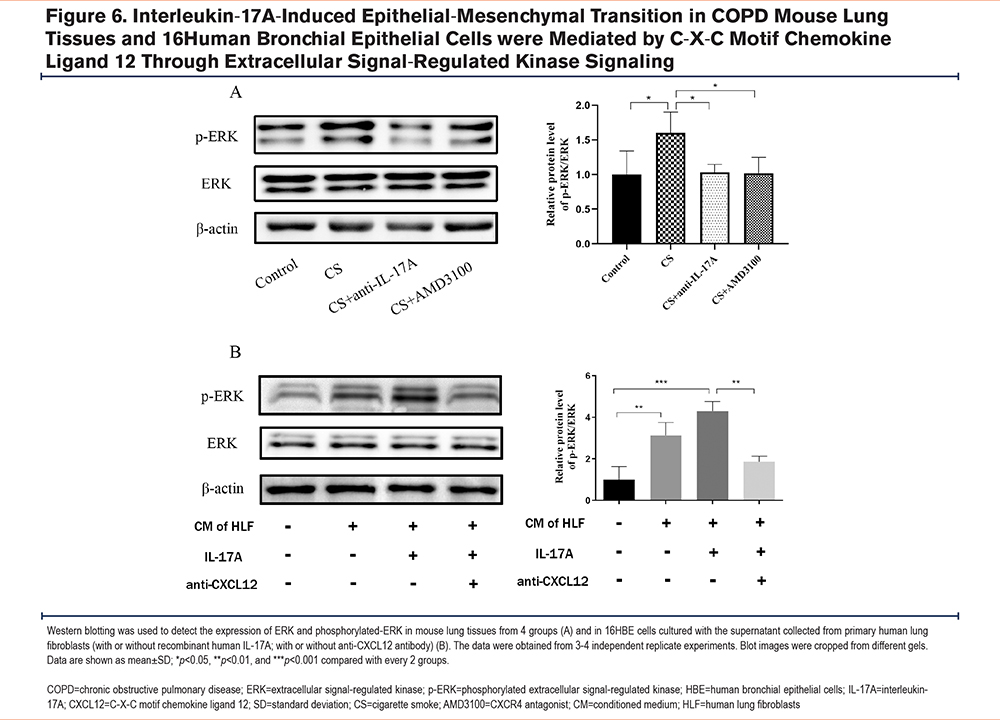
Interleukin-17A-Induced Epithelial-Mesenchymal Transition in COPD Mouse Lung Tissues and 16Human Bronchial Epithelial Cells Were Mediated by C-X-C Motif Chemokine Ligand 12 Through Extracellular Signal-Regulated Kinase Signaling
To explore the ERK-mediated signaling pathways during EMT in response to IL-17A stimulation, ERK phosphorylation levels were measured in the lung tissues of BALB/c mice. Western blotting revealed that the ratio of p-ERK/ERK was increased in CS-challenged mice, and ERK phosphorylation was decreased following treatment with AMD3100 (Figure 6A). Moreover, changes in ERK signaling were examined in 16HBE cells. The 16HBE cells cultured with the IL-17A-stimulated human fibroblast supernatant showed an increase in the p-ERK proportion, which was reduced upon CXCL12 neutralization (Figure 6B).
Discussion
Airway remodeling is a structural change characterized by thickening of airway walls, subepithelial collagen deposition, and excessive mucus secretion.20 Airway epithelial cells are the primary targets of inhaled toxic gases and particles, including CS. Chronic exposure to a repetitive environmental injury may lead to the persistent activation of pathways involved in airway epithelial repair, such as EMT, which can initiate airway remodeling.21 The interaction between constituent cells of lung tissues, particularly fibroblasts, and HBE cells plays a key role in EMT in patients with COPD. Culture supernatants derived from lung fibroblasts of patients with COPD promote EMT in healthy adult HBE cells. Consistently, it has been demonstrated that activated primary lung fibroblasts promote EMT in 16HBE cells.16
CS is a common etiologic factor for COPD, and it is associated with a series of pathophysiological changes. IL-17A is elevated in patients with COPD as well as in smokers with normal lung function. In the current study, IL-17A expression was elevated in lung tissues of mice chronically exposed to CS, particularly at 24 weeks. In a bleomycin-induced murine model of fibrosis, IL-17A significantly increased the pulmonary fibroblasts, type I collagen, and transforming growth factor-beta in vitro, which were attenuated by an anti-IL-17A antibody.22 In asthma, IL-17A can cause lung fibroblasts to overexpress α-SMA, collagen 1, and other activation markers.23 Thus, IL-17A is directly associated with fibroblast activation during chronic airway inflammation. In this study, primary lung fibroblasts were cultured from fresh lung tissues and treated with IL-17A to mimic a COPD-related inflammatory response in vitro. Our results demonstrated that fibroblasts are indeed activated by IL-17A; however, the most appropriate IL-17A stimulation concentrations are based on the different species.
Further, IL-17A-activated pulmonary fibroblasts secreted CXCL12, and CXCL12 expression was elevated in lung tissues of the COPD mouse model. Although fibroblasts are a primary source of CXCL12, the binding of CXCL12 to its receptor CXCR4 expressed on the cell surface of fibroblasts results in a positive feedback loop and is an important mechanism of cascade amplification. The CXCL12-CXCR4 pathway seems to be more mature in neoplastic diseases. Its activation usually promotes malignant tumor proliferation and metastasis,24 and it has been shown to influence over 20 malignant tumors of different origins from organs and types, such as colorectal cancer,25 hepatocellular carcinoma,26 and breast cancer.27 Additionally, the CXCL12-CXCR4 pathway plays a potential role in inflammatory diseases. The number of CXCR4+ fibrocytes is increased in the peripheral blood of patients with an acute exacerbation of COPD.15 The CXCL12-CXCR4-dependent interaction within lung tissues is involved in the development of chronic airway inflammation in COPD, which is closely related to lung function decline.15 In the present study, IL-17A and CXCL12 were inextricably linked to airway remodeling in COPD. To specifically block the CXCL12/CXCR4 pathway, AMD3100 was injected intraperitoneally into the COPD mouse model, and the results suggested an attenuation of EMT. Similar trends were also observed using antibodies in the COPD mouse model. In vitro experiments showed a trend of EMT attenuation following the treatment of 16HBE cells with a culture supernatant from anti-CXCL12-neutralized activated fibroblasts. These results strongly suggest that IL-17A affects the entry of airway epithelial cells into EMT through CXCL12, which ultimately leads to airway remodeling.
After binding to CXCR4, CXCL12 can initiate various signaling pathways, including mitogen-activated protein kinase (MAPK) and its downstream p38 and ERK1/2 protein kinases (including Akt, protein kinase, and Rac), phosphoinositide 3-kinase, Ras, and c-Jun N-terminal kinase.28,29 p-ERK is a downstream signaling protein that contributes to cascade amplification of the MAPK signaling pathway. Binding of CXCL12 to CXCR4 has been shown to stimulate the proliferation of various tumor cell lines and their migration and adhesion to extracellular matrix components by activating these downstream signal transduction pathways. For example, Lin et al demonstrated that CXCL12, which acts through CXCR4 and activates the Rac/ERK signaling pathways, could induce the expression of connective tissue growth factor in human lung fibroblasts, and potentiate their transdifferentiation into myofibroblasts.30 Wang et al demonstrated that the autocrine CXCL12/CXCR4 axis could mediate the metastatic properties of esophageal cancer stem cells and it was dependent on ERK1/2 signaling.31 Pulmonary fibrosis and metastatic progression of tumors are similar to airway remodeling and inseparable from EMT. The current study investigated ERK signaling downstream of airway remodeling in COPD. In lung tissues of the COPD mouse model, the ERK pathway was clearly activated and was partially inhibited following treatment with an anti-IL-17A antibody or AMD3100. In vitro experiments revealed a markedly activated ERK pathway in 16HBE cells cultured with the CXCL12-containing fibroblast supernatant, followed by its partial inhibition upon CXCL12 neutralization.
AMD3100, which has been approved for use by the United States Food and Drug Administration, plays an important role in mobilizing hematopoietic stem/progenitor cells and treating autoimmune diseases and asthma.32 In recent years, studies on AMD3100 have been limited to malignant tumors, such as cholangiocarcinoma, pancreatic cancer, and ovarian cancer, and they have confirmed its anti-pulmonary fibrosis effect.33 The results of the current study support the known role of AMD3100. Treatment of mice with AMD3100 was able to decrease the production of CXCR4/CXCL12 and it attenuated CS-induced COPD, despite incomplete inhibition. Barwinska et al proposed that the protective effects of AMD3100 on CS-induced chronic lung injury may be due to bone marrow mobilization that increases the availability of hematopoietic progenitor cells (HPCs) for lung cell maintenance or repair. This is particularly necessary since both the number and proliferative potential of bone marrow HPCs have been shown to drop with CS exposure.34 However, the blood or sputum levels of CXCR4-positive HPCs in humans or mice with COPD were not measured, while altered CXCL12 levels in HPCs derived from blood samples of individuals with COPD are an observational marker.
In this study, IL-17A promoted EMT progression in a mouse model of CS-induced COPD through CXCL12 derived from activated lung fibroblasts. EMT progression was associated with an interaction between epithelial cells and fibroblasts, mediated by ERK activation. These findings are related to airway remodeling in COPD, and they provide new insights into inflammatory mediators, chemokines, and cellular mechanisms that mediate EMT in lung epithelial cells. Thus, the data presented herein could help guide the exploration of novel treatment targets that would benefit the patients by reducing airway remodeling and improving their prognosis.
Conclusion
In conclusion, the current study revealed the critical role of IL-17A-activated fibroblast-derived CXCL12 in the long-term CS exposure-induced COPD mouse model, supporting IL-17A and CXCL12 as mechanistic links between CS-induced COPD and airway remodeling. These results provide novel insights into the mechanisms of COPD.
Acknowledgements
Author contributions: XC, LC, GC, and JW were in charge of data curation. XC, LC, JL, and HW were in charge of methodology. XC and LC oversaw software. XC wrote the original draft. XC and HW reviewed and edited the manuscript. WY and HW were in charge of the funding acquisition. WY led the investigation. HW conceptualized the manuscript. All authors read and approved the final version of the manuscript.
Data sharing statement: The data that support the findings of this study are available upon request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
We thank Medjaden Inc. for its assistance in the preparation of this manuscript.
Declaration of Interests
The authors declare that they have no competing interests.