Running Head: AATD in a Never Smoker With Novel Mutation
Funding Support: None
Date of Acceptance: September 12, 2024 | Publication Online Date: October 2, 2024
Abbreviations: AAT=alpha-1 antitrypsin; bkgd=background; BLVR=bronchoscopic lung volume reduction; COPD=chronic obstructive pulmonary disease; CT=computed tomography; DLCO=diffusing capacity of the lungs for carbon monoxide; DLCO/VA=diffusing capacity divided by the alveolar volume; FEF=forced expiratory flow; FEV1=forced expiratory volume in 1 second; FVC=forced vital capacity; GOLD=Global initiative for chronic Obstructive Lung Disease; PFT=pulmonary function testing; RV=residual volume; SPECT=single-photon emission computed tomography; TLC=total lung capacity; UAE=United Arab Emirates
Citation: Barjaktarevic IZ, Hong AW, Hoover A, et al. Alpha-1 antitrypsin deficiency in a young never smoker with novel Pi*Null homozygous mutation: a case report. Chronic Obstr Pulm Dis. 2024; 11(6): 624-629. doi: http://doi.org/10.15326/jcopdf.2024.0518
Background
Alpha-1 antitrypsin (AAT) is a serine protease inhibitor that inhibits neutrophil elastase.1-3 AAT deficiency results from mutations in the SERPINA1 gene, resulting in the loss of neutrophil elastase inhibition. AAT deficiency and unbalanced protease activity of neutrophil elastase leads to the lytic degradation of pulmonary connective tissues, predisposing affected individuals to the early development of chronic obstructive pulmonary disease (COPD), particularly emphysema.4 Typically, the mean age of onset of lung disease5,6in patients with a history of smoking ranges from 32 to 41 years, while never-smokers may remain asymptomatic until around the age of 48.
The most common mutations associated with AAT deficiency, such as the "Z" and "S" alleles, result in reduced levels and function of AAT protein, and in instances such as P*ZZ homozygosity, may lead to severely reduced levels below pulmonary protective levels.7Additionally, gain-of-toxic function mutations can produce misfolded, insoluble AAT protein polymers in hepatocytes, ultimately resulting in cirrhosis.7 In contrast, mutations that result in undetectable AAT protein levels, known as "Q0" or "Null" mutations, are exceedingly rare and, while not affecting the liver, may confer a significantly higher risk of emphysema due to the near absence of AAT production.8 These mutations can be caused by nonsense mutations, intron mutations, frameshift mutations, or large gene deletions, leading to either premature cessation of protein production or translation of disrupted protein, both most often leading to a functional silencing of the SERPINA1 allele.9 Since Null mutations do not induce AAT polymerization, the risk of liver disease is not present. Nevertheless, the risk of lung disease development is considerably high.8 There are over 30 reported Null AAT alleles, and this report adds another previously never reported mutation in a young homozygous, never-smoking patient with end-stage respiratory failure.
Case Presentation
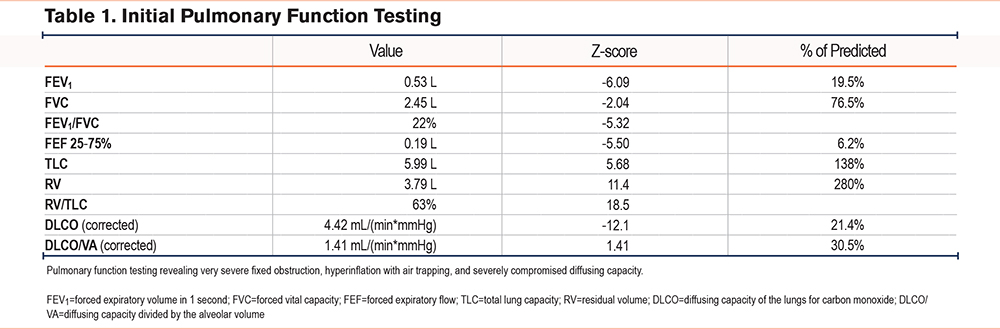
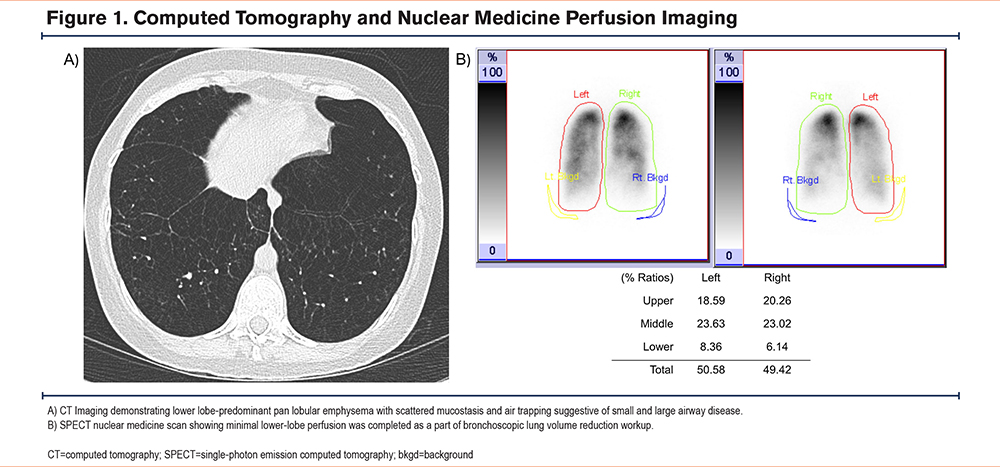
We present a 35-year-old female of Middle Eastern descent with no significant medical history, born to consanguineous parents without significant medical histories (first cousins). She was a never-smoker with moderate exposure to burning incense without a prior history of childhood respiratory illness or asthma and was diagnosed with emphysema at age 30. She first reported experiencing shortness of breath on exertion with near-syncope during travel out of the country, with her dyspnea progressing over the years to the point of limiting her activities of daily life. She was treated with a combination inhaler, which included beclometasone, formoterol, and glycopyrronium, and had no exacerbations but was started on home oxygen due to hypoxia with ambulation. By the time she presented at the international clinic in the United Arab Emirates (UAE), where she was found to have undetectable antigenic levels of AAT, her pulmonary function test demonstrated an obstruction with percent-predicted forced expiratory volume in 1 second (FEV1) 28%, total lung capacity (TLC) 105%, and diffusing capacity 30% of predicted values. At that point, she sought further evaluation for her advanced emphysema in the United States with the consideration of potential lung transplantation. On our exam, her pulmonary auscultation revealed significantly decreased breath sounds bilaterally but no wheezing. She had no resting hypoxemia, and her initial venous blood gas levels had a pH of 7.34 and partial pressure of carbon dioxide within arterial or venous blood levels of 45mmHg. Pulmonary function testing (PFT) at our institution indicated severe obstructive lung disease with an FEV1 to forced vital capacity (FVC) ratio of 22%, FEV1 of 19.5% of predicted, TLC of 138%, and a diffusing capacity of 30.5% (Table 1). High-resolution computed tomography (CT) imaging revealed diffuse panlobular emphysema affecting all lung lobes. AAT levels were undetectable (<20mg/dL; reference range: 90mg/dL to 200mg/dL or 16uM to 36.8uM) (Figure 1) and using qualitative isoelectric focusing/immunoturbidimetry phenotyping, a complete absence of AAT protein was demonstrated, suggesting the possibility of 2 Pi*Null variant mutations. Genetic sequencing by Invitae (San Francisco, California) labs analyzed the SERPINA1 gene’s transcript NM_000295.4, revealing homozygosity for a novel c.82del (p.Gln28Argfs*52) mutation on Exon 2, with consequential frameshift mutation and the generation of a premature stop codon 52 amino acids beyond the 28th position, resulting in an absent or disrupted protein product. While we have not performed any additional tests to further confirm the possibly altered function of this single-nucleotide polymorphism, this variant has not been present in the population database (gnomAD no frequency) nor reported in the literature in individuals affected with SERPINA1-related conditions. ClinVar contains an entry for this variant (Variation ID: 2155461), and based on the above, was deemed pathogenic based on Invitae's proprietary variant classification, Sherloc, a validated system based on American College of Genetics and Genomics guidelines.10,11 Finally, given the literature's conventional naming standards, this novel mutation was named after the patient's origin, leading to the proposed name of this Null variant: Q0Bani-Yas. With her diagnosis, genetic counseling was provided for both the patient and her family.
Following the guidelines of the Global initiative for chronic Obstructive Lung Disease (GOLD), we optimized the patient's conservative COPD management regimen, including optimizing delivery methods, maximizing triple inhaler regimen with adjunctive oral therapies (oral N-acetylcysteine), addressed comorbidities which included upper-airway cough syndrome and gastroesophageal reflux syndrome, and completed pulmonary rehabilitation.12 In addition, the patient was started on standard-dose plasma-derived AAT augmentation therapy, and arrangements were made to secure the continuation of the treatment upon her return to her native country. Subsequent PFTs showed a mild improvement in FEV1 to 27.3%.
While the evaluation for lung transplantation was initiated, due to the patient's reluctance, the decision was made to postpone transplantation and reevaluate in several months. Simultaneously, consideration was also given to bronchoscopic lung volume reduction (BLVR), given the presence of significant hyperinflation (residual lung volume 238%) and minimal lower-lobe perfusion based on the single-photon emission computed tomography nuclear imaging (Figure 1). An attempt at BLVR in 2023 was aborted due to the presence of significant collateral ventilation using the CHARTIS system.13 Upon conveying the patient's evaluation and treatment history in the United States to her local treatment team, she returned to the UAE. Through collaboration between our institution and the international drug delivery team, arrangements were made for the patient to receive AAT augmentation therapy in her home country based on regular shipment of the product. Nevertheless, within 6 months of her return to UAE, she continued to deteriorate with worsening hypoxemia and frequent hospitalization, which all led to the decision to proceed with a lung transplantation locally. She underwent a successful double-lung transplant and, 4 months postsurgery, has been recovering well and without significant complications. She has been off oxygen, with reported prebronchodilator spirometry without obstruction and FEV1 94% and FVC 82% of predicted values, on standard immunosuppression and antibiotic prophylaxis. No AAT augmentation was continued post-transplant.
Discussion and Conclusions
This case report details the progression of severe end-stage lung disease in a young, never-smoker female with a novel Null SERPINA1 homozygous mutation variant. It is relevant for these reasons: (1) we report a novel, to our knowledge, never-reported mutation of SERPINA1, Q0Bani-Yas, in its homozygous form, and (2) our report highlights the potential for emphysema to manifest in its devastating form even earlier in patients with undetectable levels compared to those with AAT mutations causing deficiency.1
Although more than 200 SERPINA1 mutations have been documented, AAT deficiency remains frequently underdiagnosed, leading to delayed diagnosis and exacerbated symptom severity.14,15 The current guidelines from the American Thoracic Society and the European Respiratory Society recommend AAT deficiency testing for individuals exhibiting early-onset emphysema (age 45 years or younger) or emphysema in the absence of established risk factors, a recommendation consistent with the presentation of our patient.6 Initial diagnostic steps typically involve assessing serum AAT levels and genotyping, primarily targeting the most prevalent S and Z alleles.16 However, more extensive workup may be occasionally required to identify specific, rare SERPINA1 gene variants, which may lead to distinct disease manifestations. For example, certain well-documented mutations, such as the MMalton variant, are associated with both emphysema and liver disease.14 Other mutations, such as α1-antitrypsin Pittsburgh (α1-AT-P), produce dysfunctional protein, which acts as a potent thrombin inhibitor, potentially leading to fatal hemorrhagic diathesis.17 In contrast, Null-variants, like the one identified in our patient, do not elevate the risk of liver disease since they avert the production of misfolded protein in hepatocytes but result in no detectable levels of AAT and are strongly correlated with the development of emphysema, possibly to a higher degree in comparison to most classic forms of AAT deficiency, Pi*ZZ.18 While our case of a never-smoker with devastating lung disease at a relatively young age may support this hypothesis, strong data comparing the risk toward COPD between severely AAT-deficient and absent phenotypes is largely missing, which is not unusual as rare SERPINA1 variants are most often present in heterozygous forms and are more frequently undiagnosed. In rare instances of Pi*null homozygosity, the consanguinity of parents may be the key predisposing factor that can lead to the unfortunate combination of abnormal genes; thus, careful attention to the patient's family history in cases of rare genetic disease is always advisable.19 For rare SERPINA1 variants such as null-alleles, conventional diagnostic methods such as isoelectric focusing or genotyping may prove insufficient, necessitating specialized sequencing techniques such as next-generation sequencing.14 For example, a recently discovered null mutation Q0Bolton generates truncated AAT protein from a null variant.20 From this, it could be possible that our mutation, Q0Bani-yas, may have the propensity to transcribe highly unstable small proteins that could not be detected by our standard quantitative methods and may not mean that there were truly undetectable levels of AAT protein as our measurements suggested. Thus, future investigations with this particular SNP may warrant the use of more sensitive studies, such as elastase complex formation immunosorbent assays.21 While this particular exploration was not within the scope of this first report of this mutation, the discovery of future null mutations should prompt functional assay testing of the particular null mutation.
In addition to the above, our approach to the diagnosis and management merits certain discussion. Although, due to ongoing management, we cannot report the ultimate outcomes, this report exemplifies the need for a comprehensive approach to severely diseased AAT patients,16 which, in our case, included simultaneous AAT augmentation therapy, lung transplant evaluation, and BLVR evaluation. AAT augmentation based on weekly intravenous infusions of plasma-derived AAT is recommended and associated with a decreased rate of lung function decline and survival,22 and data suggest that the lack of access to intravenous AAT replacement therapy could hinder these survival benefits.23 The potential detrimental effects of lack of access to treatment highlight the importance of global AAT networks and large international cohorts24-26and associations with global reach,27 which can facilitate the diagnosis and management of AAT deficiency in countries without access to testing and treatment. BLVR has been one of novel nonpharmacologic approaches in severe emphysema. BLVR may reduce hyperinflation and improve quality of life, potentially deferring lung transplantation, and has recently been gaining traction in AAT-deficient patients with severe COPD.28,29 Unfortunately, despite adequate clinical features qualifying the patient for valve placement, which included fissure completeness, systematic evaluation during bronchoscopy revealed the presence of collateral ventilation, resulting in aborting the plan for BLVR in this case.13 Lung transplant represents the ultimate resource for patients with end-stage respiratory failure due to AAT deficiency. It is characterized by higher survival rates but also a higher risk of common post lung transplant complications in patients with AAT deficiency COPD versus non-AAT deficiency COPD.30
Acknowledgements
Author contributions: IB and AWH were responsible for drafting major manuscript revisions, while IB also served as principal investigator. SN and AH provided insight into the genetic counseling aspects. In addition to revisions, MB lent his expertise in the molecular genomics of alpha-1 antitrypsin deficiency. All authors assisted with editing and review of the manuscript.
We would like to thank the patient and her family for allowing us to report this case. We acknowledge Invitae Corporation of San Francisco, California, for their assistance in sequencing the novel allele. We would also like to acknowledge Dr. Stanley Nelson for his assistance with genetic counseling and thank the patient’s local pulmonologist, Dr. Said Isse, from Cleveland Clinic in Abu Dhabi, UAE.
Declaration of Interests
There are no financial conflicts related to this work to disclose.