Running Head: The Alpha-1 Biomarkers Consortium Study
Funding Support: This work was supported by the National Institutes of Health UG3/UH3 HL152323 and the Alpha-1 Foundation.
Date of Acceptance: April 17, 2025 | Published Online: April 24, 2025
Abbreviations: A1BC=Alpha-1 Biomarkers Consortium; A1F=Alpha-1 Foundation; AAT=alpha-1 antitrypsin; AATD=alpha-1 antitrypsin deficiency; AcPGP=acetylated proline-glycine-proline; AECOPD=acute exacerbation of COPD; AUDIT-C=Alcohol Use Disorder Identification Test; BCSS=Breathlessness, Cough, and Sputum Scale; CAT=COPD Assessment Test; CC-16=club (clara) cell secretory protein 16; CLDQ=Chronic Liver Disease Questionnaire; COPD=chronic obstructive pulmonary disease; COPDGene®=COPD Genetic Epidemiology study; CRP=C-reactive protein; CT=computed tomography; EARCO=European Alpha-1 Research Collaboration; ECLIPSE=Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints; FEV1=forced expiratory volume in 1 second; iPSCs=inducible pluripotent stem cells; MMPs=matrix metalloproteinases; mMRC=modified Medical Research Council; NIH=National Institutes of Health; OSMB=observational safety monitoring board; PBMCs=peripheral blood mononuclear cells; Perc15=15th percentile Hounsfield Unit value; REDCap=Research Electronic Data Capture; SGRQ=St George’s Respiratory Questionnaire; SOBQ=University of California San Diego, Shortness of Breath Questionnaire; SP-D=surfactant protein D
Citation: Goldklang MP, Pirozzi C, Barjaktarevic I, et al. Rationale and design of the alpha-1 biomarkers consortium study. Chronic Obstr Pulm Dis. 2025; 12(4): 274-284. doi: http://doi.org/10.15326/jcopdf.2025.0603
Online Supplemental Material: Read Online Supplemental Material (340KB)
Introduction
Alpha-1 antitrypsin deficiency (AATD) is the most common genetic cause of chronic obstructive pulmonary disease (COPD). Although AATD is regarded as a classical Mendelian disorder, marked variability in the development and severity of disease-associated phenotypes exists.1-3 This variability underscores the need for an improved characterization of the range of clinical outcomes associated with specific disease-causing mutations and the influence of additional modifying genes and environmental exposures on disease presentation. To date, the National Heart, Lung, and Blood Institute Registry of AATD,4,5 the QUANTitative lung computed tomography UnMasking emphysema progression in AATD (QUANTUM-1),6 and the control arms of some comparative studies,7-9 are the largest longitudinal studies of patients with AATD. These studies were performed before widespread quantitative computed tomography (CT) scans for the measurement of emphysema and prior to the adoption of triple therapy in the management of COPD and reflect historical smoking rates in the U.S. population. Contemporaneously, the European Alpha-1 Research Collaboration (EARCO) International Registry10,11has recruited a larger number of patients with AATD in Europe, with extensive analyses regarding clinical characterization and blood biomarkers, but the study contains only qualitative, not quantitative measures of emphysema, in only a subset of patients. Furthermore, augmentation therapy is not widely available in all countries within the EARCO registry. Therefore, there is still much to be defined regarding the current natural history of, treatment strategies for, and the clinical course of AATD for patients in the United States.
Pathophysiology of AATD
Based on population studies,12,13 it is estimated that 1 in 2800 to 1 in 5000 individuals in the United States has a severely deficient genetic variant on both SERPINA1 sites (ZZ, or rare null alleles), extrapolating to between 67,000 and 117,000 severely deficient patients. Alpha-1 antitrypsin (AAT) is a serine protease inhibitor that inactivates neutrophil elastase and matrix metalloproteinases to maintain the protease-antiprotease balance in the lung. Individuals with AATD inherit variant alleles of SERPINA1 that cause misfolding of the AAT protein within the hepatocyte, with resultant low levels of functional plasma AAT.14,15 This misfolded protein triggers an endoplasmic reticulum stress response in the hepatocyte, with resultant liver inflammation and fibrosis.16 Initial interest in imbalances between proteases and their endogenous inhibitors stemmed from the observation of an increased incidence of emphysema in smokers with AATD.17-19 Neutrophil elastase is well known to be a major protease involved in tissue destruction of emphysema20,21 and AAT is a key inhibitor of elastase, with its functional loss resulting in damage of the extracellular matrix.
Computed Tomography Imaging in AATD
Unlike the general COPD population that can enroll large numbers of participants to test and validate new therapies based on spirometric measures,22,23 the limited patient pool in AATD creates a challenge. Previously, CT-based measures of emphysema and lung density have been utilized in AATD clinical trials. The RAPID7 and RAPID-OLE24 trials tested the efficacy of weekly AAT augmentation therapy on altering changes in CT lung density (e.g., the 15th percentile Hounsfield Unit value [Perc15]) in 180 participants and to date have established Perc15 as the most widely accepted biomarker of lung destruction in AATD. Recent advances in CT image postprocessing techniques now facilitate the estimation of lung tissue biomechanics through paired inspiratory and expiratory high-resolution chest CT scans. The Jacobian determinant is a measure of local volume change and substantially explains differences between density-based measures of emphysema and the degree of airflow obstruction on spirometry,25,26 but has not been evaluated in AATD. Furthermore, complex branching patterns of the airways and subtle variations in these patterns due to disease presence can be quantified using the airway fractal dimension,27 which has been independently associated with respiratory quality of life, functional capacity, exacerbations, lung function decline, and mortality,27 again never evaluated in AATD. This study presents unique opportunities for exploring CT imaging metrics to better understand disease severity and progression. The identification of novel imaging-based biomarkers with clinical and/or pathological relevance has the potential to accelerate the pipeline of therapies available to this vulnerable patient population.
Endpoints in AATD Clinical Trials
General COPD studies have focused on endpoints such as change in forced expiratory volume in 1 second (FEV1),28 but require large numbers of patients for statistical significance, given variation in FEV1 over time. Interventional clinical trials require large numbers of patients (in some cases over 1000) to demonstrate an effect on exacerbation rate.22,29 In AATD, this approach is infeasible due to a limited patient pool. The AATD clinical research community has, therefore, turned to methods including change in CT lung density as a surrogate marker of lung destruction in clinical trials, including the evaluation of the clinical efficacy of AAT augmentation therapy.7,24 The identification of additional blood or imaging biomarkers with clinical or pathological relevance as meaningful intermediate endpoints that may decrease sample size and/or trial length has the potential to improve the pipeline of therapies available to this patient population.
Methods
Study Objectives
The goal of the Alpha-1 Biomarkers Consortium (A1BC) is to better understand the heterogeneity of AATD and associate blood or imaging biomarkers with specific disease phenotypes that might be applied to characterize disease severity or predict clinical outcomes. Biomarkers have been defined by a National Institutes of Health (NIH) working group as a “characteristic that is objectively measured and evaluated as an indicator of normal biological processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention.”30 In the A1BC, biomarkers are broadly defined to include CT-based measurements of emphysema, serum, plasma, and sputum biomarkers of inflammation and lung destruction, alongside outcomes such as spirometry and quality of life questionnaires. The study further aims to determine if there are modifying genetic relationships in the disease progression of AATD. Sequencing of the SERPINA1 gene will be performed, including in the promoter and enhancer region, on all A1BC patients to test the hypothesis that genotype-phenotype correlations exist among PiZZ AATD patients.
Study Design
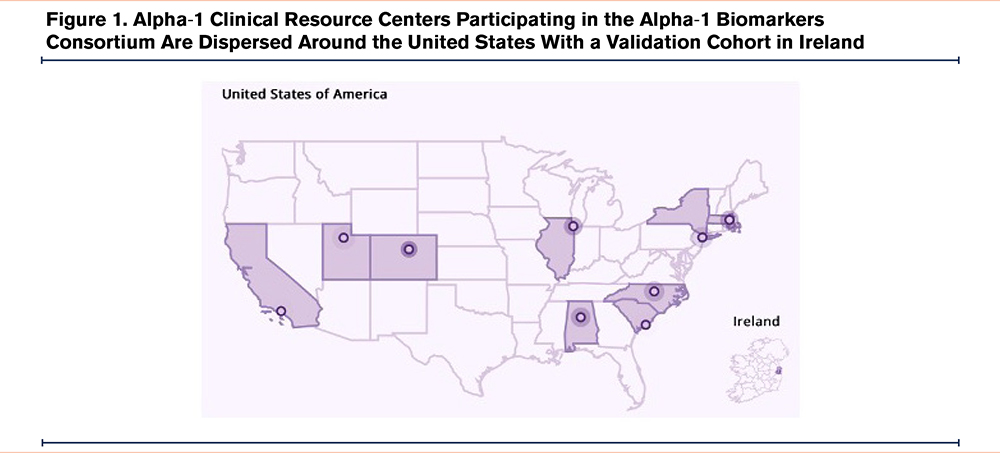
In 2019, the Alpha-1 Foundation (A1F) established a research registry (A1F Registry, NCT04157049) housed within the A1F containing over 1200 discrete data including contact information and extensive patient-reported and medical record-verified clinical data. The first phase of this study was to establish a clinical cohort of alpha-1 patients with the methodology to link A1BC participants to the A1F Research Registry. All self-reported PiZZ AATD participants in the A1F Registry, 18 years or older, were invited to join the A1BC, with a final enrollment goal of 270 participants. A total of 9 A1F clinical resource centers will be recruiting geographically dispersed U.S. patients (Table 1 and Figure 1). A separate validation cohort in Ireland has been established. The study design was approved by the Western Institutional Review Board-Copernicus Group, and both the A1F Registry (NCT04157049) and the A1BC (NCT05297812) are enrolled in clinicaltrials.gov. A1F Registry and A1BC registration numbers will be linked for data sharing. After completion of the study, patient data and biomaterials will be transferred to the A1F Registry and will be used in accordance with A1F’s mission.

Liberal inclusion and exclusion criteria have been selected to ensure that the entire range of AATD pulmonary manifestations in the PiZZ cohort is included (Appendix Table 1 in the online supplement). Inclusion criteria are largely based on genotype, adult age (owing to the low incidence of emphysema in children and radiation risk exposure over a lifetime), and a willingness to share data with the A1F. Participants will be included regardless of augmentation therapy status, with data collection regarding dose and interval collected on relevant patients. Crossover of augmentation therapy status during the study will be collected. Patients will be excluded if listed for a lung or liver transplant. Since CT metrics of emphysema serve as the primary endpoint of this study, patients with diagnoses that could cause alterations in CT imaging were specifically excluded from the cohort; such exclusions have been implemented in other trials utilizing CT densitometry, including in the COPD Genetic Epidemiology (COPDGene®) study.31 Therefore, patients with major thoracic surgeries, lung volume reduction procedures, and automatic internal cardiac defibrillators and similar devices were excluded. Additionally, patients with clinically significant bronchiectasis were excluded.31 While patients cannot be enrolled in therapeutic clinical trials at the time of A1BC consent and first visit, to not hamper enrollment in the current and upcoming clinical trials across the AATD landscape, the A1BC does not prohibit enrollment in interventional clinical studies after the A1BC initial visit. At each 6-month call, patients are queried regarding clinical trial enrollment, and if newly enrolled in additional clinical trials, the clinicaltrials.gov reference number is collected.
In-person visits with blood, spirometry with and without bronchodilators, and CT imaging will occur at baseline, 18 months, and 36 months. Optional induced sputum will be performed on appropriate patients at participating centers (only 3 of 8 centers due to COVID-19 restrictions). Extensive patient-reported outcome measures will be administered. Participants will be contacted by telephone every 6 months to update clinical history, obtain current medication lists, and assess for any adverse events. Patients will be invited to complete monthly surveys of exacerbation history. A Whatman 903 blood spot card and fingerstick lancet is provided to all participants at baseline, and participants will be instructed on self-collection of blood on a dried blood spot card at the day of onset, day 3, and day 7 of exacerbations. A schedule of procedures is provided in Appendix Table 2 in the online supplement.
Measurements
Questionnaires
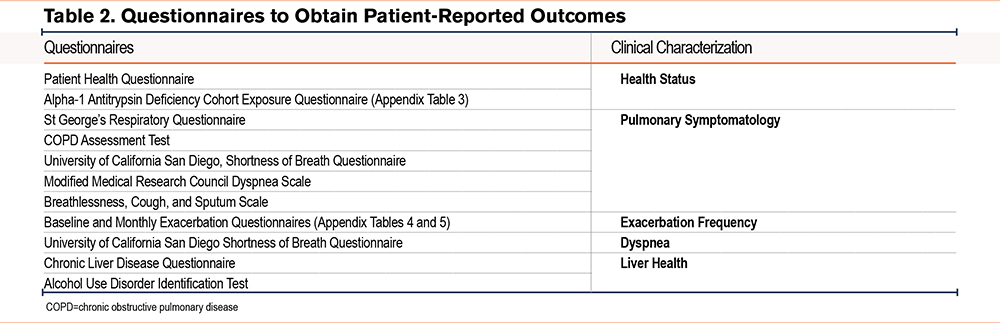
This cohort will undergo extensive analysis, including clinical characterization of exposure history and health status (Patient Health Questionnaire, A1BC Exposure Questionnaire). Table 2 lists the questionnaires used to capture clinical manifestations of lung and liver disease associated with AATD; more details are available in the online supplement.
In addition to the above, extensive AATD-related clinical questions are obtained regarding diagnosis, including method of diagnosis, familial index case, and duration between symptom onset and diagnosis. Information regarding childhood or adult asthma, bronchiectasis, and NTM infection is obtained. Specific information about augmentation dosing, intervals, and location of administration will be collected. Other nonpulmonary AATD-related clinical findings are directly queried, including neonatal and childhood liver disease, liver fibrosis and cirrhosis, vasculitis, and panniculitis. Clinician-obtained and questionnaire-based information regarding smoking history and alcohol use will be collected.
Computed Tomography Imaging
All participants enrolled in the study will undergo acquisitions of high-resolution CT scans of the chest at total lung capacity and residual volume at the baseline visit and subsequently at 18 months and 36 months of follow-up. The acquisition protocol will be standardized, and dose adjusted according to body mass index using the SubPopulations and InteRmediate Outcome Measures In COPD Study chest CT protocol.32 Subsequent CT scans will be acquired using the same scanner and the same protocol for each participant at each site, unless there is a significant change in body weight. The COPDGene phantom will be used to calibrate CT scanners for quality assurance.33 Digital Imaging and Communications in Medicine standard images will be transferred to the University of Alabama at Birmingham Lung Imaging Laboratory using a Health Insurance Portability and Accountability Act-compliant secure web portal system.32
Nasal Transcriptome
Based on the united airway disease hypothesis, which proposes that specific inflammatory process within the respiratory tract manifests in both the upper and lower tracts,34 we hypothesized that the nasal transcriptome will be a minimally invasive “window” to view the pathobiology of AATD-related respiratory disease development. Study participants will undergo nasal swab sampling of each nare prior to sputum induction for RNA sequencing and nasal transcriptome analysis. We plan to evaluate the potential of nasal swab-based sampling to reflect the immunological and inflammatory patterns associated with the development of lung disease and aim to compare the nasal to peripheral blood transcriptome patterns.
Plasma and Serum Biomarkers
Identification and validation of biomarkers in AATD, especially those present in blood, has remained a challenge. While the Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) study and other studies have shown differences in lung-derived and liver-derived biomarkers between patients with COPD and smoking controls,35-43 and that some of these markers have been associated with disease severity (fibrinogen and C-reactive protein [CRP]),44,45 rate of decline in lung function (club [clara] cell secretory protein 16 [CC-16]), exacerbation frequency (surfactant protein D [SP-D] and CRP),38,44,46-49 and risk of mortality (pulmonary and activation-regulated chemokine/chemokine ligand 18),41,50 they excluded patients with PiZZ AATD. Furthermore, although soluble receptor for advanced glycation end products and SP-D are associated with loss of lung tissue as assessed by CT lung density, these biomarkers are understudied in AATD-associated emphysema. Therefore, these biomarkers need to be assessed in individuals with AATD to clarify whether the associations observed in usual COPD are maintained in this rare condition. Of particular interest will be the ability of the ECLIPSE biomarkers to correlate with clinical outcome measures, including rate of loss of CT lung density, as well as their ability to identify patients with frequent exacerbator and/or rapid decliner phenotypes. The optimal technique to measure blood biomarkers that associate with CT density, the hallmark of AAT lung disease, is an unbiased proteomic approach. While the A1BC may be underpowered for such analyses, an unbiased proteomic analysis will be performed on all A1BC serum samples.
Central to the present investigation is the concept of a loss of balance between pro-inflammatory and anti-inflammatory mediators and between proteases and antiproteases in AATD, and multiple potential biomarkers reflective of this derangement will be evaluated. Collagen breakdown by prolyl endopeptidases produces the biologically active extracellular matrix peptide acetylated proline-glycine-proline (AcPGP), a potent chemokine associated with neutrophilic inflammation in cigarette smoke-mediated emphysema development.51-55 In addition to neutrophilic inflammation and elastase activity, it is important to note other proteinases have been implicated in COPD pathogenesis; cathepsins and matrix metalloproteinases (MMPs) play important roles in the protease imbalance in human emphysema.56-64 Therefore, AcPGPs and levels of MMPs in the circulating blood will be evaluated.
Peripheral Blood Mononuclear Cell Banking and Induced Pluripotent Stem Cells
Patient-derived inducible pluripotent stem cells (iPSCs) allow disease modeling in differentiated cells that contain the genome of the patient from whom they were derived, including any disease-relevant variants. In recent years, iPSC models have been applied to the study of AATD-associated hepatocyte injury and have both recapitulated known disease features and extended understanding of cellular injury mechanisms.65-69 Peripheral blood mononuclear cells (PBMCs) that can be used to generate iPSCs will be isolated from A1BC study participants and cryopreserved as live cells for banking in an established PBMC/iPSC repository.70,71 Specific participant samples with distinct clinical phenotypes or novel genetic variants will be identified for iPSC generation and differentiation to disease-relevant lineages based on phenotypes observed in study data.
Genetic Testing and Detection of Single Nucleotide Polymorphisms
Recent advances in sequencing have led to the description of new mutations in both the exon and intron portions of the SERPINA1 gene.72 It has been hypothesized that mutations in the promoter regions can account for changes in the acute phase response and subsequent increase in AAT during infection. The functional effects of polymorphisms within both the intron and promoter regions are unknown, and it is possible that there are mutations within SERPINA1 to account for the phenotypic differences between patients with the PiZZ allele.
Data Management and Analysis
Columbia University and the A1F have established a Research Electronic Data Capture (REDCap) data entry system. REDCap is a public-sourced database, which was a requirement of the NIH study.73 The REDCap for A1BC has linkage numbers with the A1F Registry to connect current data with historical clinical data.
Statistical Considerations
The goal for powering this study was to assure that longitudinal CT density changes would be seen over 3 years. Two recent studies have used 180 participants over 2 years in a 2-arm therapeutic trial (RAPID) and 330 participants over 3 years in a 3-arm therapeutic trial (Study of ProlAstin-c Randomized Therapy with Alpha-1 Augmentation). Since our study is not a clinical trial and individuals with normal lung function are invited for participation, the number of 270 participants to follow over 3 years was chosen as a pragmatic goal that was also based on the number of PiZZ patients at participating sites. While the number of patients required to observe clinical and biochemical effects in augmentation naïve patients and/or those stable on augmentation therapy will be higher than the aforementioned AATD proof-of-concept studies, we expect that this number is again covered by the proposed cohort size. In RAPID, evaluation of 54 patients from the placebo arm was sufficient to detect a significant change in desmosine/isodesmosine at 24 months.7,74 Since large prospective studies correlating inflammatory biomarkers with clinical outcomes and quantitative CT metrics of emphysema in AATD are lacking, this will be the largest prospective biomarker study ever conducted in AATD.
Association of Predictors With Clinical Outcomes
Marginal association analysis will be conducted using linear regression models by regressing the CT measurement of Perc15 on each of the K (K= 28~30 in total) blood biomarkers and lung function parameters (FEV1 percentage predicted using Global Lung Function Initiative “other” equations and FEV1/forced vital capacity ratio), adjusted for important baseline covariates including ever smoking (100 cigarettes in a lifetime), a categorical variable of 10 pack years of smoking, baseline CRP, age, and sex. We will utilize different sets of baseline covariates for sensitivity analysis, including smoking pack years. To correct for multiple testing, we plan to use both Bonferroni correction to control for family-wise error rate and use q-value to control for false discovery rate. To account for possible correlations among biomarkers, we will use the elastic-net penalized regression models with 5-fold cross-validation to select important biomarkers jointly related to the baseline Perc15 using the R package glmnet. Moreover, we will apply the weighted correlation network analysis method using the R package WGCNA to discover clusters (modules) of highly correlated biomarkers that are associated with Perc15. For each cluster consisting of multiple biomarkers, we will use kernel machine regression to capture complex nonlinear and interaction effects of multiple biomarkers associated with Perc15. Any missing data will be imputed using the multiple imputation technique implemented in the R package mice. We will begin with cross-sectional analysis for data collected at each of the 3 visits and will use linear mixed models for the repeated measurements of all 3 visits to assess any longitudinal change over time. Important covariates from repeated measurement analysis, including the number and severity of pulmonary exacerbations, will be explored.
Findings will be validated by a companion cohort of participants from the National Alpha-1 Registry of Ireland, based at the Irish Centre for Genetic Lung Disease (Beaumont Hospital/Royal College of Surgeons in Ireland, Dublin). Methods for validation will be modeled to explore significant findings from the U.S. cohort, and discrepancies will be probed to determine the differences in covariates between the 2 cohorts.
Genetic Analyses
With sequencing data of the SERPINA1 gene, we will first perform standard quality control and data preprocessing. SERPINA1 sequencing will be performed on the entire gene, with promoter region, exon, and introns included. Then, we will perform genetic association analysis using a linear regression model for Perc15 measured at each visit and linear mixed models for the 3 repeatedly measured Perc15 to identify putative causal genetic variants, adjusted for sex, age, and smoking. Gene-environment analysis will be performed with the inclusion of an interaction term between genetic variants and important environmental variables (e.g., cigarette smoking). Additionally, we will also perform genetic interaction analysis by including interaction terms between genetic variants to explore possible epistatic effects.
Study Oversight
An observational safety monitoring board (OSMB) has been established to include 2 pulmonologists and one statistician. The OSMB meets approximately once every 6 months to review patient enrollment, safety related to the study procedures, and adverse events. The open OSMB meeting is attended by NIH and A1F representatives. Adverse events are defined primarily as related to study procedures. Special events of clinical interest will be captured, including AECOPDs, decompensated liver disease, and enrollment in clinical trials. Furthermore, incidental findings primarily related to lung nodules on CT scans will be collected.
Potential Outcomes and Conclusions
The above-described study will be the largest cohort of AATD patients with PiZZ genotype prospectively assembled, longitudinally followed, and systematically analyzed with regard to biomarkers of lung disease. The characterization of a full complement of biomarkers, including blood biomarkers, CT imaging biomarkers, patient-reported outcomes, exposure history, lung function, and genomics in a cohort of individuals with AATD has never been done at this large scale in a longitudinal fashion. The evaluation of associations between these characteristics and clinical outcomes in cross-sectional analysis, as well as over time in the 3-year longitudinal follow-up, will improve understanding of the heterogeneity of AATD and identify factors associated with disease severity and progression. The results have the potential to improve care for individuals with AATD by identifying individuals at risk for more severe lung or liver disease manifestations earlier, before deterioration, and may lead to future interventions that will improve clinical outcomes.
Acknowledgements
Author contributions: MPG, CP, IB, SBh, MBD, NGM, OJM, JMW, AW, CS, and JMD conceived the study and developed the theoretical framework. All authors contributed to and approved the final manuscript.
The authors wish to thank Drs. Antonello Punturieri and Lisa Viviano from the National Institutes of Health and the Alpha-1 Foundation leadership for their support of this consortium.
Alpha-1 Biomarker Consortium Study Group
Columbia University: Jeanine M. D’Armiento, Monica P. Goldklang, Zhonghua Liu, Laura D. Fonseca, Tina Zelonina, Alexis C. Fisher, Niki Trivedi, Sydney Harris, Sabrina Palumbo, Kiran K. Chada, Devipriya Harinath, Sanjiv Prasad
University of Utah: Cheryl Pirozzi, Cassie E. Larsen, Alexis G. Vanwagoner
University of California, Los Angeles: Igor Barjaktarevic, Russell Buhr, John Belperio, Semi Yoon, Vyacheslav Palchevskiy, Lung Wing
University of Alabama at Birmingham: Mike Wells, Surya P. Bhatt, Sandeep Bodduluri, Abhiraj Pudhota
University of North Carolina at Chapel Hill: M. Bradley Drummond, Caleb C. Hemphill, Kelsey B. Haywood, Walker A. Long
University of Chicago: D. Kyle Hogarth, Vanita Patel
Alpha-1 Foundation: Randel Plant, Alison Keaveny, Nadine Nuchovich, Jennifer Illarramendi, Laura D. Fonseca, Charles Emala, Jon Hagstrom, Scott Santarella
Royal College of Surgeons in Ireland: Noel G. McElvaney, Oliver J. McElvaney, Suzanne M. Roche, Daniel D. Fraughen
National Jewish Health: Robert A. Sandhaus, Ariana K. Talaie
Boston University: Andrew Wilson, Joseph E. Kaserman, Mark Dodge, Shumin Guan
Medical University of South Carolina: Charlie Strange, Leah Benn, Allison Burton, Gwen Hayden, Chloe Jacobs, Kristen Neff, Melinda M. Talley, Andrea J. Swartz
Declaration of Interest
In the last 36 months, the following disclosures were provided.
MPG has contracted research support for clinical trials from Sanofi, Arrowhead, NovoNordisc, Takeda, InhibRx, Mereo, Vertex, and Grifols; research grant support from the Alpha-1 Foundation, and has been paid fees for advisory work from Takeda, GSK, Sanofi, Korro, Inhibrx, Bridge Bio, and Grifols, and received support for medical writing from Takeda.
CSP has contracted research support for clinical trials from Inhibrx, Vertex, and AstraZeneca; has received consulting fees from Takeda, and has received honoraria for lectures from Medscape, Advancing Knowledge in Healthcare, the Academy for Continued Healthcare Learning, Medscape Education, the Cleveland Clinic, the Alpha-1 Foundation, and Projects in Knowledge.
IB has contracted research support for clinical trials from Theravance and Viatris, Aerogen, Takeda, Amgen, the Alpha-1 Foundation, and Johnny Carson’s Foundation, and has received consulting fees from AstraZeneca, Sanofi, Regeneron, Grifols, Verona Pharma, Inhibrx, Takeda, Genentech, Aerogen, Theravance, and Viatris.
SBh has received consulting fees from Sanofi, Regeneron, GSK, Genentech, Boehringer Ingelheim, Apreo, AstraZeneca, Chiesi, Verona, and Merck; has received honoraria for lectures from MedScape, IntegrityCE, Integratis Communications, Illuminate Help, and Horizon CME.
MBD has received grants to his institution for research by PCORI, Vertex, Teva Pharmaceuticals, the American Lung Association, Boehringer-Ingelheim, Midmark, Inc, and the National Institutes of Health (NIH); has received consulting fees from AstraZeneca, Verona, Takdea, Becker Pharma, GSK, Stratos, Genentech, and Amgen, and serves on the medical and scientific advisory boards for the COPD Foundation and the Alpha-1 Foundation.
DKH has received honoraria for lectures for Grifols, Takeda, and Sanofi, and has received consulting fees from Advanced Infusion Care and Wave Life Sciences.
NGM has received grants for investigator-initiated studies from Grifols and the Alpha-1 Foundation, consulting fees from CSL Behring, BEAM Therapeutics, Intellia Therapeutics, and GSK, and support to attend meetings from Grifols.
OJM has received grants from the Cystic Fibrosis Foundation and the University of Washington, consulting fees from Grifols, and serves on the scientific advisory committee for Grifols.
RP has received honoraria for participation in the Takeda Educational Materials Working Group.
RS has received grant support from Inhibrx and Sanofi, consulting fees from Grifols, CSL Behring, Takeda, Korobio, Beam, Wave, and Biomarin with payments all payments directed to the not-for-profit disease management organization AlphaNet; has a pending patent through his institution for CT analysis software, is a data safety monitoring board member for Takeda, Beam, and Biomarin, and has medical director roles in the Alpha-1 Foundation, AlphaNet, and AlphaNet Canada.
JMW has clinical trial support from the NIH, Veterans Administration, ARCUS-Med, Medscape, Verona Pharma, Grifols, the Alpha-1 Foundation, Inhibrx, and the American Lung Association; has a patent with Mereo BioPharma, has received advisory board fees from AstraZeneca, Takeda, GSK, Bavarian Nordic, Krystal Biotech, Sanofi, and Verona Pharma, and has received support for medical writing from Takeda, GSK, and Verona Pharma.
AW has received consulting fees from Takeda and is the scientific director of the Alpha-1 Foundation.
CS has grants paid to the Medical University of South Carolina from the Alpha-1 Foundation, Beam, Biomarin, Grifols, Krystal, Mereo, and Takeda, and is a consultant for CSL Behring, GSK, Sanofi, and Takeda.
JMD has grants and contracts for clinical trials from Mereo, Takeda, Arrowhead, Vertex, and Inhibrx, has received consulting fees from Sanofi and Bridge Bio, and is participating in advisory boards for Sanofi and Takeda.
SBo, LF, AK, ZL, NN, and SP have no disclosures.