Running Head: Relationship Between Circulating Cathepsin Levels and COPD
Funding Support: This study was supported by the Health Department of Jilin Province (2023LC008).
Date of Acceptance: July 26, 2025 | Published Online: August 6, 2025
Abbreviations: CI=confidence interval; COPD=chronic obstructive pulmonary disease; ECM=extracellular matrix; GWAS=genome-wide association study; IV=instrumental variable; IVW=inverse-variance weighted; LASSO=least absolute shrinkage and selection operator; MR=Mendelian randomization; MVMR=multivariable MR; PRESSO=pleiotropy residual sum and outlier; PTMs=post-translational modifications; RCT=randomized controlled trial; SNP=single nucleotide polymorphism
Citation: Duan C, Zhang A, Tian S. Genetic evidence for causal relationships between circulating cathepsin levels and chronic obstructive pulmonary disease: a Mendelian randomization study. Chronic Obstr Pulm Dis. 2025; 12(5): 380-389. doi: http://doi.org/10.15326/jcopdf.2025.0626
Online Supplemental Material: Read Online Supplemental Material (480KB)
Introduction
Chronic obstructive pulmonary disease (COPD) is a progressive inflammatory lung disorder characterized by persistent airflow limitation.1,2 The disease clinically manifests through respiratory symptoms including dyspnea, chronic cough, excessive sputum production, and wheezing.2 Pathologically, COPD encompasses 2 principal components: emphysema, involving alveolar sac destruction leading to impaired gas exchange, and chronic bronchitis, featuring persistent bronchial inflammation with mucus hypersecretion.3 The alveolar tissue degeneration in emphysema reduces lung elasticity, while chronic bronchitis causes airway narrowing through inflammatory thickening and mucus plugging. Despite therapeutic advances, COPD remains incurable, with treatment focusing primarily on symptom management and disease progression delay.1 These clinical realities underscore the critical importance of early detection and sustained intervention, which can substantially improve patient outcomes and quality of life.
Cathepsins are a family of proteolytic enzymes predominantly found in all animals in addition to other organisms. There are several types of cathepsins, each categorized mainly by their enzymatic nature and substrate specificity, including cathepsins B, D, L, S, K, G, H, V, and C among others.4 These enzymes are known for their roles in lysosomal degradation, where they contribute to the breakdown of proteins inside the lysosomes, critical cellular organelles responsible for digesting various biomolecules. Their broad range of functions in cellular maintenance and regulation renders specific cathepsins crucial for cellular functioning and highlights their potential roles in the management and targeted treatments of various complex diseases.5-7
Growing evidence implicates cathepsins in COPD development through multiple pathological mechanisms.8 Cathepsins S, L, and K contribute to disease progression by degrading extracellular matrix (ECM) components, including elastin and collagen, in lung tissue. This proteolytic activity drives the structural remodeling of airways and parenchyma characteristic of COPD.9-13 Beyond ECM degradation, cathepsins participate in COPD-associated inflammation by activating cytokines and chemokines that sustain pulmonary inflammatory cascades. Their pathogenic role is further supported by elevated levels detected in COPD patients’ sputum and bronchoalveolar lavage fluid.13,14 Cathepsins also regulate immune responses frequently dysregulated in COPD. For example, cathepsin G modulates neutrophil function10 – a critical defense mechanism in lungs. Abnormal cathepsin G activity may impair neutrophil responses, exacerbating inflammation and tissue damage.8 Furthermore, increased cathepsin E protein in the lung epithelium of COPD patients has been observed.15
While these observations derive primarily from in vitro and observational studies (which are susceptible to confounding and reverse causality), they collectively suggest cathepsins as potential therapeutic targets. However, the causal relationship between cathepsin levels and COPD risk requires validation through robust methods.
Mendelian randomization (MR) is a research method used in epidemiology to assess the causal relationship between potentially modifiable risk factors and health outcomes.15 This technique leverages genetic variation as a surrogate to examine the causal effect of a modifiable exposure on disease in observational data. In other words, it uses genetic variants as instrumental variables (IVs) to estimate the causal effect of an exposure on the outcome.16,17 MR relies on the natural random assortment of genes at conception, which obeys Mendel’s laws of inheritance. This allocation mimics the randomization process in a randomized controlled trial (RCT), minimizing confounding factors that typically affect observational studies. While MR studies cannot replace RCTs, they offer insights that help bridge the gap between correlation and causation. In this study, we aimed to elucidate the causal relationship between cathepsin levels and COPD risk using MR analyses, and, therefore, to provide valuable insights on the prevention and early intervention for COPD.
Methods and Materials
Experimental Data
Genetic association statistics for 9 cathepsin (i.e., cathepsin B, E, F, G, H, O, L2, S, and Z) levels were derived from the INTERVAL study, comprising 3301 participants of European ancestry.18 COPD summary statistics were obtained from the FinnGen consortium,19 including 6915 COPD cases and 186,723 controls. Lastly, we used a summary genome-wide association study (GWAS) data for smoking from the Medical Research Council-Integrative Epidemiology Unit consortium,20 which included 280,508 cases and 180,558 controls. Ethical approval for this study was waived by the First Hospital of Jilin University Institutional Review Board as no original research data were collected.
IVs for cathepsin levels were selected based on the following criteria: a linkage disequilibrium threshold of R2<0.001 within a 10,000kb clumping window, and a genome-wide significance level of 5 × 10−6.
Mendelian Randomization
For a single nucleotide polymorphism (SNP) to serve as a valid IV in MR analyses, 3 fundamental assumptions must be satisfied: (1) relevance assumption dictates that the SNP must be strongly correlated with the exposure, (2) independence assumption requires that the SNP must be independent of any confounders that affect the relationship between the exposure and the outcome, and (3) exclusion restriction assumption requires that the SNP should not have a direct association with the outcome, nor should it be related to the outcome through any pathways other than the exposure.
The inverse-variance weighted (IVW) method served as our primary analytical approach for estimating causal effects, offering optimal statistical power when all genetic variants meet IV assumptions.21 However, recognizing that violations of these assumptions—particularly through horizontal pleiotropy—could bias IVW estimates, we implemented supplementary pleiotropy-robust methods to validate our findings. These included MR-Egger regression, which accounts for balanced pleiotropy22; the weighted median approach, providing consistent estimates when even up to 50% of weights derive from invalid instruments23; the weighted mode method, effective when the largest SNP cluster shares a common causal estimate24; and MR-pleiotropy residual sum and outlier (PRESSO), which identifies and corrects for outlier variants.25
Both the MR-PRESSO global test and MR-Egger intercept (MR-Egger intercept p-value < 0.05) were used to identify outliers and detect horizontal pleiotropy. Additionally, the MR-PRESSO outlier test was conducted to mitigate or eliminate horizontal pleiotropy by removing outliers (p-value of the MR-PRESSO global test <0.05). Cochran’s Q test was used to assess heterogeneity among SNPs, with a p-value of <0.05 indicating heterogeneity. If the p-value was less than 0.05, a random-effect model was used to estimate causal effect size. Otherwise, a fixed-effect model was applied instead. The R TwoSampleMR package26 was utilized for conducting 2-sample MR analyses. The MR-PRESSO tests were carried out using the MR-PRESSO package.
Reverse MR analyses in which COPD was considered as the exposure and cathepsins as the outcome were performed to explore reverse causality, using the GWAS studies from the forward MR analyses. Next, multivariable IVW MR analysis involving 9 cathepsins as predictors was conducted using the R MendelianRandomization package. To address multicolinearity, least absolute shrinkage and selection operator (LASSO) analysis was performed to select relevant features and construct the final model, and the R MrLasso package27 was employed for this analysis. Conditional F-statistics were calculated using the R multivariable MR (MVMR) package.
Lastly, posthoc statistical power assessment of the MR analysis was conducted using the mRnd online tool.28,29 The current study was designed following the Strengthening the Reporting of Observational Studies in Epidemiology-MR checklist.30
Results
Univariable Mendelian Randomization
The causal relationship between 9 cathepsins (cathepsin B, E, F, G, H, O, L2, S, and Z) and the risk of COPD was investigated using a 2-sample univariable MR analysis. Initially, all p-values from Cochran’s Q statistics exceeded 0.05. In conjunction with the results from leave-one-out plots (Supplementary Figure 1 in the online supplement), it was concluded that significant heterogeneity was absent. Furthermore, the MR-Egger intercept test revealed no horizontal pleiotropy, with a p-value greater than 0.05. All F-statistics of single SNPs were larger than 10.
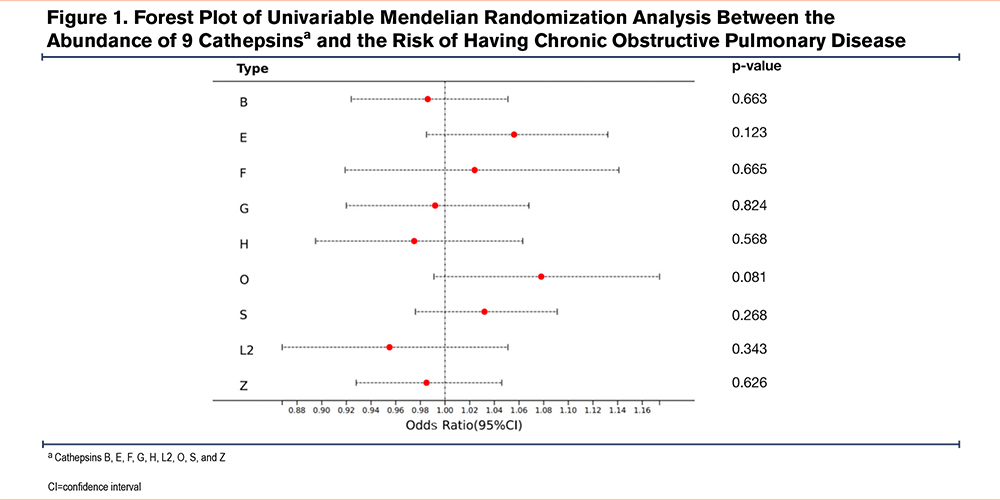
The results of univariable MR analysis are tabulated in Table 1, while the forest plot in Figure 1 graphically elucidates IVW results. For example, our analysis showed that the risk of developing COPD for a one-unit increment in abundance level of cethepsin O was estimated to be 1.078 (odds ratio [OR]=1.078, p=0.081, 95% confidence interval [CI]=0.991–1.174) by the IVW method. All 4 complementary MR methods supported in concordant with this null relationship. The OR was estimated as 1.076 (p=0.478, 95% CI=0.885–1.309) by MR-Egger, 1.033 (p=0.596, 95% CI=0.917–1.163) by weighted median, 1.011 (p=0.900, 95% CI=0.861–1.187) by weighted mode, and 1.044 (p=0.401, 95% CI=0.948–1.149) by MR-PRESSO. Regarding cathepsin S, which has been reported to associate with the development and progression of COPD by numerous prior studies, our analysis found a nonsignificant positive causal effect of this protein on COPD risk. Specifically, OR was estimated as 1.037 (p=0.464, 95% CI=0.940–1.145) by IVW, 1.064 (p=0.556, 95% CI=0.869–1.302) by MR-Egger, 1.132 (p=0.053, 95% CI=0.999–1.282) by weighted median, 1.140 (p=0.150, 95% CI=0.959–1.356) by weighted mode, and 1.037 (p=0.471, 95% CI=0.989–1.145) by MR-PRESSO, respectively. Overall, none of these 9 cathepsins were found to cause an increase or decrease in developing COPD. Furthermore, 4 MR methods comparison plots are shown in Supplementary Figure 2 in the online supplement, demonstrating robust MR results.
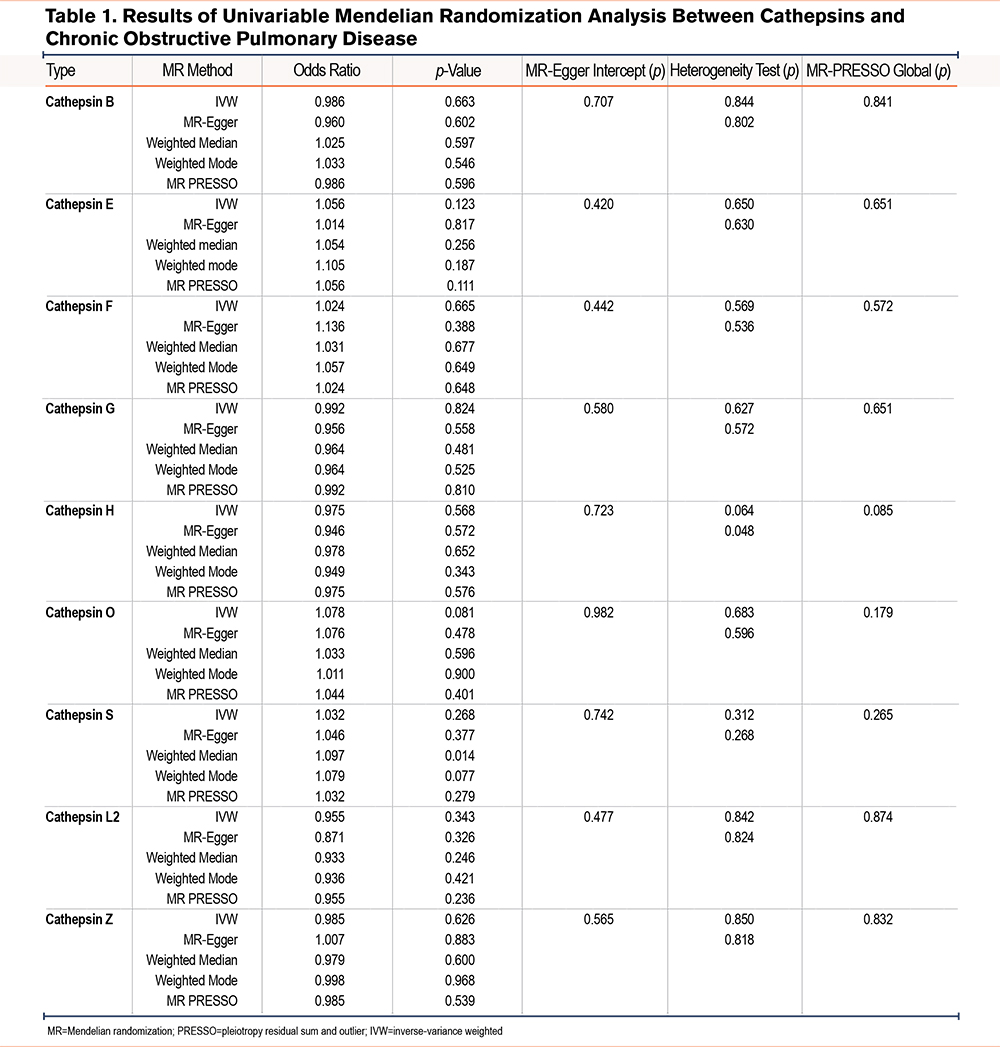
Subsequently, reverse MR analysis was performed. Null reverse causal relationship was identified between 9 cathepsin types and the risk of developing COPD, which were supported consistently by all MR methods based on corresponding adjusted p-values. For these MR analyses, neither heterogeneity (Cochran’s Q p-value >0.05) nor horizontal pleiotropy (MR-Egger intercept p-value >0.05) was detected. The reverse MR results are shown in Supplementary Table 1 in the online supplement.
Multivariable Mendelian Randomization
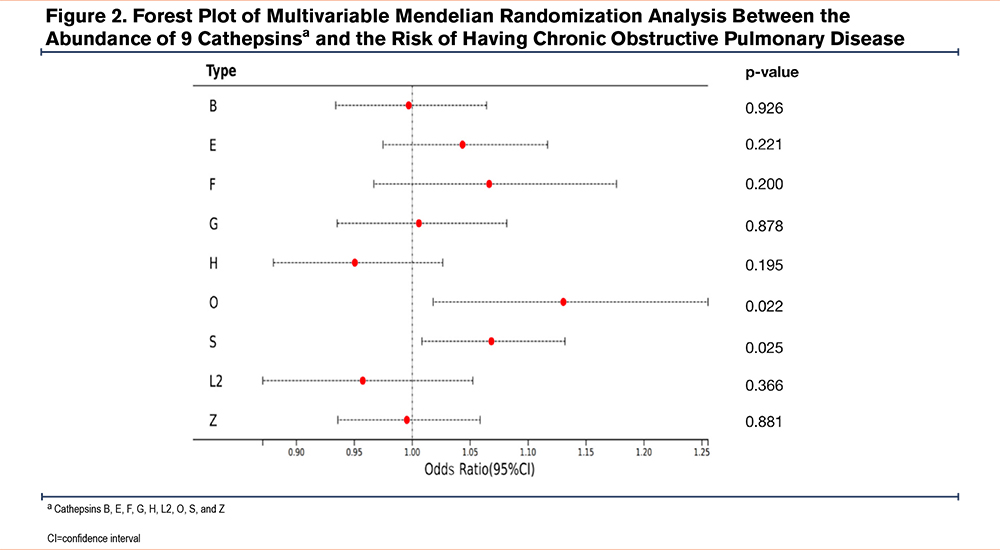
MVMR analyses (including IVW, MR-Egger, and MR-PRESSO methods) were performed to analyze the genetic predisposition for 9 cathepsin types in relation to the risk of having COPD, which identified both cathepins O and S as a risk factor for COPD (Table 2). Specifically, OR was estimated as 1.130 (p=0.022, 95% CI=1.018–1.255) for cathepsin O and 1.068 (p=0.025, 95% CI=1.008–1.132) for cathepsin S by IVW, respectively. The forest plot in Figure 2 graphically elucidates MVMR IVW analysis results. To address potential multicolinearity between cathepin types and weak instrument bias (some cathespin types have a small conditional F-statistic), we performed MR LASSO analysis to select more highly related subtypes, which identified cathepins B, O, S, and Z out of 9 cathepins. Based on these 4 cathepins, we redid MVMR analyses (Table 3) and the calculated conditional F-statistics for these 4 cathepins indicated marginal weak instrument bias. The results are in line with MVMR analysis result with all 9 cathepsin types as covariates.
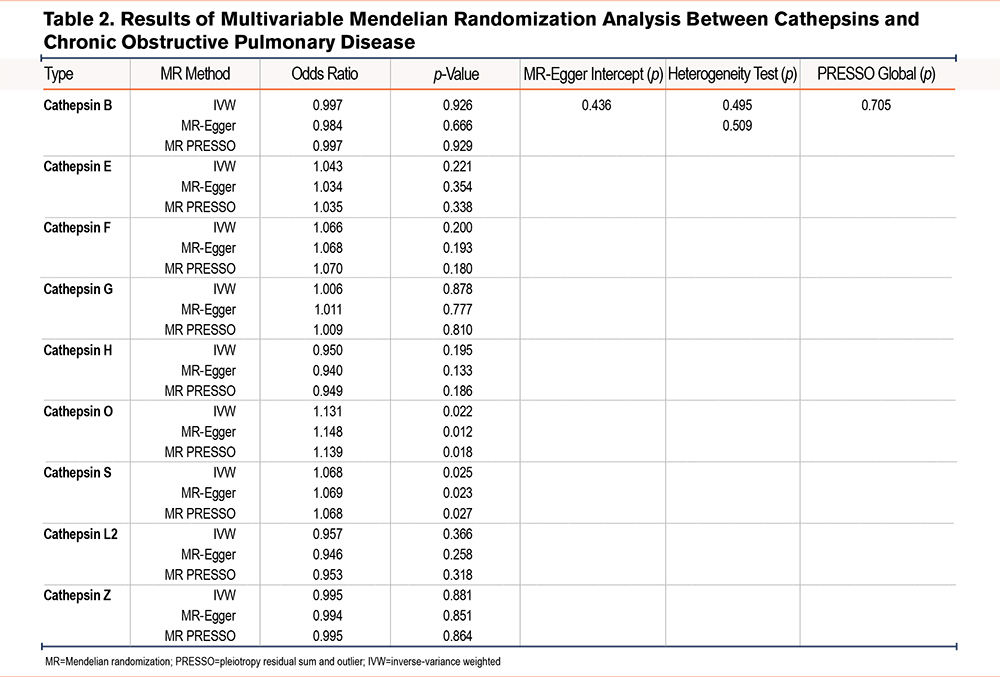
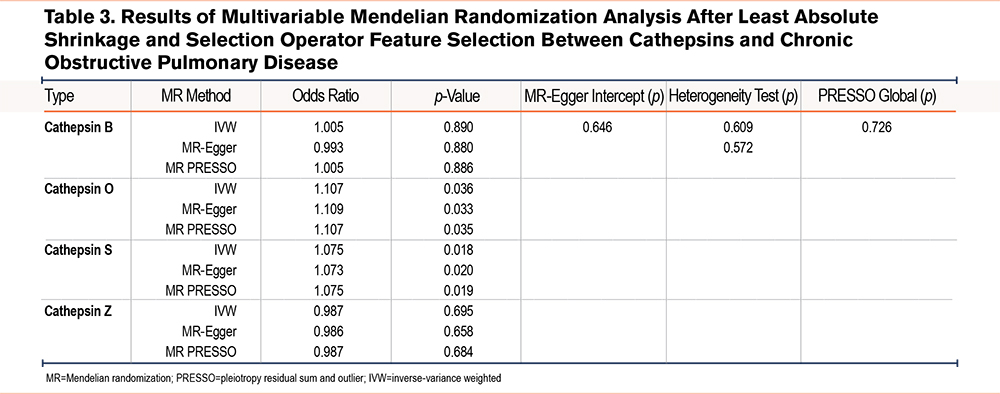
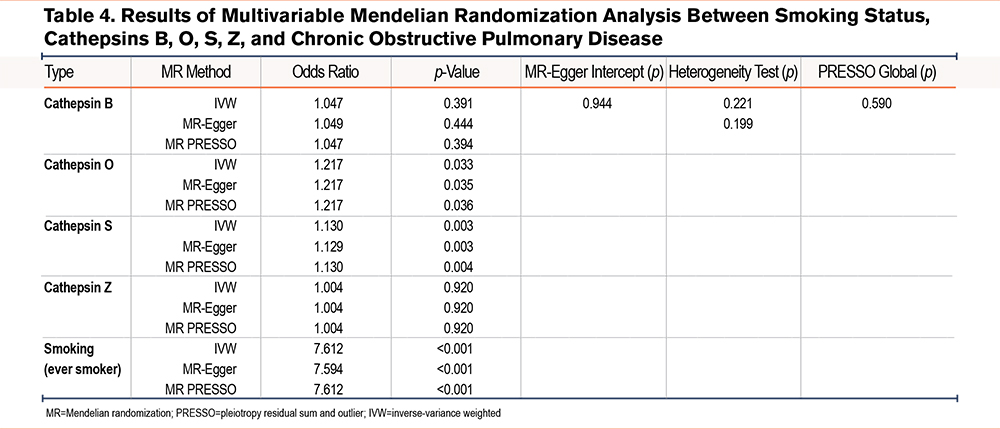
Furthermore, given that COPD is a heavily smoking-mediated disease, we also performed a MVMR using smoking and cathepins B, O, S, and Z as covariates. The analysis results indicated that after adjusting for smoking status, both cathepsins O and S were genetically related to COPD risk. Specifically, OR was estimated as 1.217 (p=0.033, 95% CI =1.096–1.458) for cathepsin O and 1.130 (p=0.003, 95% CI=1.043–1.224) for cathepsin S while OR for smoking was as 7.614 (p<0.001, 95% CI =3.819–15.180) by IVW, respectively (Table 4).
Posthoc Power Calculation
Lastly, a posthoc power calculation was performed to assess the statistical power of the current MR study. At a significance level of 0.05, a sample size of 193,638 individuals including 6915 cases (3.57%) exhibited a power of 0.80 and 0.56 to detect a 1.2- (OR=1.2) and 1.15-fold risk (OR=1.15) of developing COPD per one-unit increase in the genetically predicted specific cathepsin level. Here, we assumed that these SNPs accounted for approximately 3% of the variance in the cathepsin levels. The sample size of the current MR study was still inadequate to detect a subtle effect.
Discussion
In this study, we employed a comprehensive MR approach incorporating univariable, reverse, and multivariable analyses to investigate potential causal relationships between cathepsin levels and COPD risk. Our MVMR results identified cathepsins O and S as potential risk factors for COPD. Existing evidence suggests cathepsin O contributes to COPD pathology through several interconnected pathways. As a protease capable of degrading ECM components, cathepsin O may promote the tissue remodeling and destruction characteristic of COPD. This is supported by observations of elevated cathepsin O levels in COPD patient lung tissues compared to healthy controls.31 The enzyme's ECM-degrading activity, particularly targeting structural proteins in lung tissue, likely contributes to emphysema development - a hallmark feature of COPD involving alveolar wall destruction that impairs lung elasticity and gas exchange. Beyond its structural effects, cathepsin O appears to influence COPD progression through inflammatory modulation. The enzyme regulates cytokine and chemokine activity, potentially exacerbating the chronic inflammation that drives COPD pathogenesis. Furthermore, emerging evidence positions cathepsin O as a responder to oxidative stress,32 a key driver of COPD development in individuals exposed to cigarette smoke and environmental pollutants. In this context, cathepsin O may participate in processing damaged proteins and organelles resulting from oxidative stress in COPD patients.
Cathepsin S, another key member of the cathepsin protease family, has been consistently shown to be elevated in both lung tissues and serum of COPD patients compared to healthy individuals.7,33 As a potent protease, cathepsin S efficiently degrades critical ECM components including elastin and collagen - structural proteins essential for maintaining normal lung architecture.34 The excessive proteolytic activity of overexpressed cathepsin S may drive the pathological tissue destruction characteristic of emphysema, a defining feature of COPD that involves alveolar wall breakdown and progressive loss of lung elasticity. Beyond its direct effects on parenchymal destruction, cathepsin S-mediated ECM degradation likely contributes to the pathological airway remodeling observed in COPD. By altering the composition and integrity of airway connective tissue, cathepsin S may promote structural changes that lead to airway narrowing and increased stiffness.9-12,35 These alterations can significantly worsen airflow limitation and contribute to disease progression. Together with cathepsin O, cathepsin S appears to play a multifaceted role in COPD pathogenesis through interconnected mechanisms involving ECM degradation, inflammatory modulation, tissue remodeling, and oxidative stress responses.36-38 Their combined actions may create a self-perpetuating cycle of tissue damage and functional decline that characterizes COPD development and progression.
Several limitations should be considered when interpreting our findings. First, the relatively small sample size of the exposure GWAS required us to adopt a more lenient genetic significance threshold. Although this enabled the inclusion of additional SNPs, it may have increased susceptibility to weak instrument bias and horizontal pleiotropy. While our comprehensive sensitivity analyses helped mitigate these concerns, residual pleiotropic effects could still influence the results, as is inherent in all MR studies. Second, our analysis was constrained by the availability of GWAS summary data, which included only 9 cathepsins and missed several major cathepsin types, including cathepsins A, C, D, K, L, and W. Notably, we were unable to evaluate several cathepsins implicated in COPD pathogenesis by previous research, for example, cathepsins D and C.14,31,39 This limitation not only restricts the comprehensiveness of our investigation but may also introduce biases in the MVMR analysis results, as these omitted cathepsins could potentially confound the observed relationships. Another key limitation is that our findings derive from European populations, potentially limiting their applicability to other ethnic groups. Future research must validate these associations in diverse cohorts to determine their broader relevance. Additionally, while MR LASSO was applied to address multicollinearity, the potential for residual collinearity remains. Lastly, while MR identifies genetic associations between cathepsins and COPD, it cannot assess tissue-specific post-translational modifications (PTMs) regulating cathepsin activation, smoke-induced epigenetic and post-translational regulation, extracellular versus intracellular cathepsin activity differences, and redox modifications altering protease function. Therefore, future studies should combine proteomics, redox biochemistry, and single-cell analyses to fully elucidate how PTMs and environmental factors (e.g., smoking) modulate cathepsin-driven COPD pathogenesis.
Conclusion
To our knowledge, this study represents the first MR study to systematically investigate the causal relationship between circulating cathepsin levels and COPD risk. Our findings suggest that elevated levels of cathepsins O and S may serve as independent risk factors for COPD development, though these observations require further validation.
These findings may provide a novel therapeutic direction for COPD management through targeted modulation of specific cathepsin pathways. However, further investigation - particularly through RCTs - will be crucial to confirm these causal associations and evaluate the clinical potential of cathepsin-focused interventions for COPD patients.
Acknowledgements
Author contributions: ST conceived and designed the experiment. ST and CD ran the analysis and verified the underlying data. CD and ST wrote the original manuscript. AZ, CD, and ST were involved in data interpretation. All authors have read and approved the final version of the manuscript.
Data sharing statement: The GWAS data of cathepsins, smoking, and COPD were downloaded from the MRC Integrative Epidemiology Unit Open GWAS Project (https://gwas.mrcieu.ac.uk). The corresponding GWAS ID numbers are prob-a-718, prob-a-720, prob-a-721, prob-a-723, prob-a-724, prob-a-726, prob-a-727, prob-a-728, prob-a-729, ukb-b-20261, and finn-b-J10_COPD.
Declaration of Interest
The authors have no conflicts of interest to declare.